Page History
...
Służy do generowania tzw. plików cube programu Gaussian. Niezbędne jest dostarczenie (np. z przestrzeni storage'owej użytkownika) wygenerowanego wcześniej (np. w ogólnym eksperymencie InSilicoLab) pliku checkpoint z przechowanymi danymi obliczeń.
TeraChem
Eksperyment ten ułatwia użycie programu TeraChem służącego do wykonywania obliczeń kwantowochemicznych na kartach graficznych ogólnego przeznaczenia (GP GPU computing). Pozwala to na znaczne, nawet kilkunastokrotne przyspieszenie niektórych obliczeń (w zależności od rozmiaru układu i stosowanej metody).
| Anchor | ||||
|---|---|---|---|---|
|
Aby skorzystać z usługi InSilicoLab for Chemistry należy aktywować ją w Portalu PL-Grid. Jest ona dostępna w katalogu usług w dziale "Platforma dziedzinowa: chemia".
Dodatkowo, do wykonywania obliczeń kwantowochemicznych, wymagane jest aktywowanie usługi "Globalny dostęp gLite" dostępnej w katalogu usług w dziale "Usługi globalne". Aktywowanie tej usługi może się odbyć wyłącznie po zarejestrowaniu conajmniej jednego certyfikatu osobistego w w Portalu PL-Grid - informację na temat rejestracji oraz oraz możliwości uzyskania certyfikatu certyfikatu można znaleźć w rozdziale rozdziale "Aplikowanie, rejestracja i użycie certyfikatu" niniejszego podręcznika.
Uwaga: Zaraz po aktywowaniu usługi "Globalny dostęp gLite" informacje na temat konta użytkownika muszą zostać przekazane do infrastruktury - ten proces może trwać maksymalnie do 6 godzin. W tym czasie może nie być dostępna pełna funkcjonalność eksperymentów kwantowochemicznych (uruchamianie eksperymentów oraz pobieranie danych). Po spropagowaniu wszystkich danych pełna funkcjonalność systemu jest dostępna.
Informacje dotyczące usług i ich aktywowania można znaleźć w rozdziale "Usługi" niniejszego podręcznika.
Pierwsze kroki
Należy połączyć się z serwerem usługi
http://insilicolab.chemia.plgrid.pl/
Po wejściu do usługi, otwiera się ekran. Jeśli nie byliśmy wcześniej zalogowani do usługi, dostępny jest dla nas jedynie ekran anonimowego użytkownika. Logowanie do własnego konta następuje po przyciśnięciu przycisku Log in... i jest możliwe jest na dwa sposoby:
- Logowanie poprzez certyfikat zarejestrowany w przeglądarce użytkownika (nasze dane zostaną sczytane z certyfikatu)
- Logowanie poprzez OpenID dla infrastruktury PL-Grid. W tym wypadku zostaniemy przekierowani na stronę OpenID dla PL-Grid i poproszeni o wpisanie danych dostępowych do infrastruktury - nazwy użytkownika i hasła (tych samych które podajemy w Portalu PL-Grid) lub sczytany zostanie certyfikat umieszczony w przeglądarce. Jeśli usługa InSilicoLab for Chemistry nie była aktywna wcześniej zostanie ona aktywowana automatycznie po zaakceptowaniu odpowiedniego komunikatu. Po zalogowaniu zostaniemy automatycznie przekierowani z powrotem na stronę usługi.
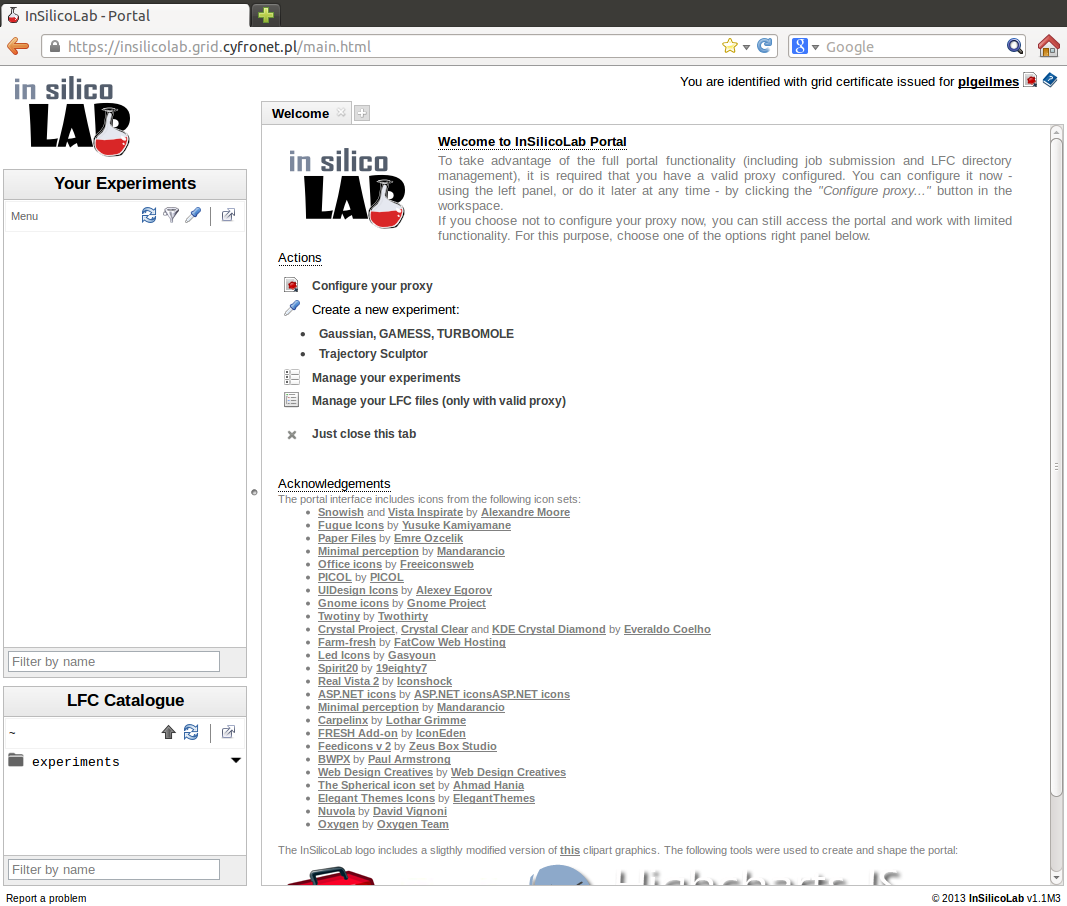
W celu przeprowadzenia obliczeń niezbędne będzie skonfigurowanie odpowiedniego certyfikatu proxy przez kliknięcie na link Configure your proxy:
Certyfikat proxy konieczny jest do przeprowadzania obliczeń z wykorzystaniem infrastruktury PL-Grid a także przeglądania danych zgromadzonych w zasobach tej infrastruktury. Nie jest natomiast konieczny do przeglądania konta użytkownika i zapisanych przez niego analiz a także do wstępnego przygotowania danych przy pomocy narzędzia Trajectory Sculptor (patrz rozdział "Zaawansowane użycie").
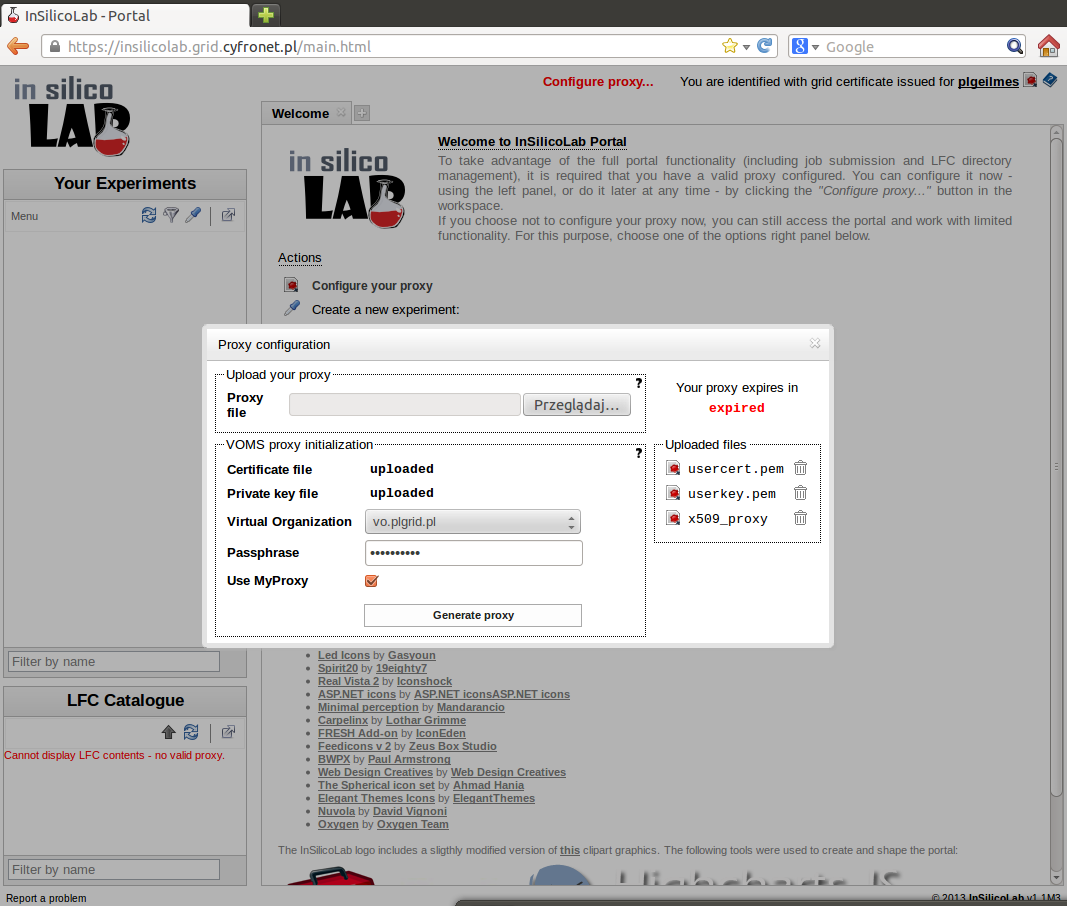
Istnieją dwie możliwości konfiguracji proxy w portalu:
- Załadowanie istniejącego proxy - wygenerowanego uprzednio na infrastrukturze. Instrukcja jak wygenerować takie proxy dostępna jest po kliknięciu w pytajnik w polu Upload your proxy. Jest to opcja bezpieczniejsza dla użytkownika, ale wymaga każdorazowego ładowania proxy po upływie jego ważności (12 godzin).
- Generacja proxy z poziomu portalu. Aby wygenerować proxy w portalu, należy załadować do niego pliki certyfikatu i klucza w formacie PEM (najczęściej są to usercert.pem i userkey.pem) lub PKCS12 (np. usercert.p12). Jest to czynność jednorazowa, i nie będzie potrzeby ponawiania jej w przyszłości jeśli certyfikat użytkownika nie ulegnie zmianie. Następnie należy wybrać wirtualną organizację - dla infrastruktury PL-Grid jest to vo.plgrid.pl, podać hasło do załadowanego klucza (UWAGA: hasło nie jest przechowywane nigdzie w systemie) oraz kliknąć przycisk Generate proxy. Zaznaczenie opcji Use MyProxy da nam możliwość automatycznego przedłużania certyfikatu proxy dla uruchomionych zadań do 168. Dodatkowe instrukcje (m. in. instrukcje postępowania w przypadku kiedy certyfikat użytkownika jest w formacie innym niż PEM) dostępne są po kliknięciu w pytajnik w polu VOMS proxy initialization.
W obu przypadkach, jeśli proxy zostało skonfigurowane poprawnie, zostanie wyświetlona na zielono ilość pozostałego czasu ważności proxy w portalu (w polu Your proxy expires in).
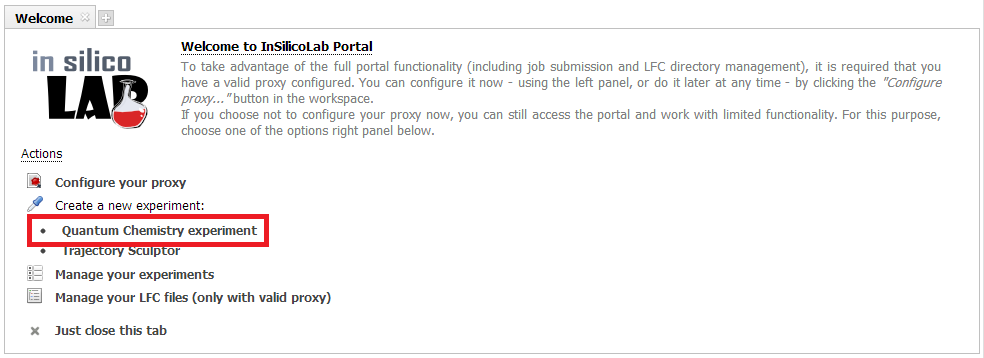
Po skonfigurowaniu certyfikatu proxy można przystąpić do właściwego eksperymentu. W celu wykonania obliczeń kwantowochemicznych z menu Create a new experiment należy wybrać pozycję Quantum Chemistry experiment.
W przeglądarce otworzy się karta nowego eksperymentu obliczeniowego wykorzystującego programu chemii kwantowej.
Uwaga: przy wyborze odpowiedniego programu chemii kwantowej, a także przy uzupełnianiu jego parametrów, można skorzystać z usługi Quantum Chemistry Advisor, agregującej dane o tych programach i wspomagającej wybór odpowiedniego do danych obliczeń.
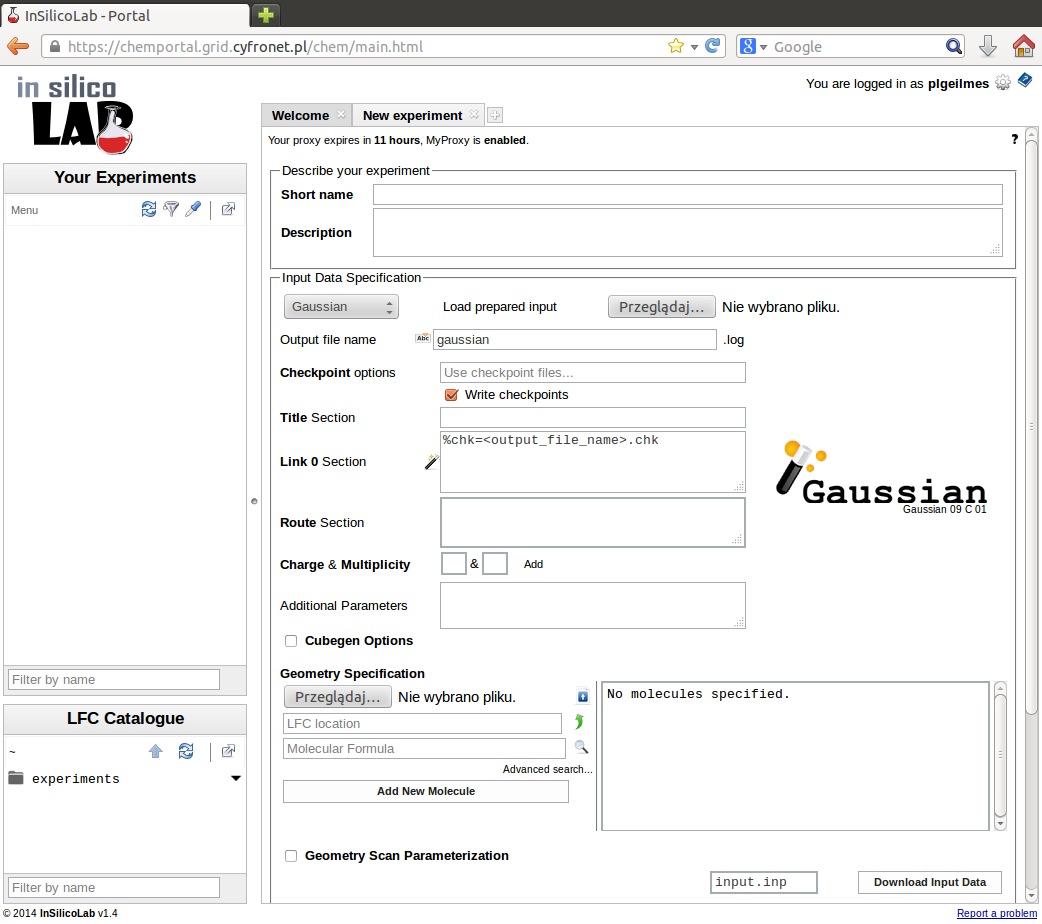
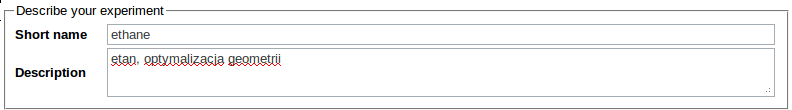
Identyfikację eksperymentu wprowadzamy podając jego krótką nazwę oraz (opcjonalnie) dłuższy opis. W przypadku niepodania krótkiej nazwy, wykorzystane zostanie pole Title formatki aplikacji kwantowochemicznej. Jeśli oba pola zostaną puste, jako krótka nazwa użyty zostanie tekst "(no title)".
Prostą metodą zadania geometrii badanego układu jest wprowadzenie jej przez załadowanie przygotowanego wcześniej pliku w Geometry Specification a następnie określenie jego typu (np. XYZ) w otwierającym się oknie menu. Domyślnie typ pliku ustawiany jest na podstawie jego rozszerzenia. Przyciskiem Finish kończymy wprowadzanie geometrii układu.
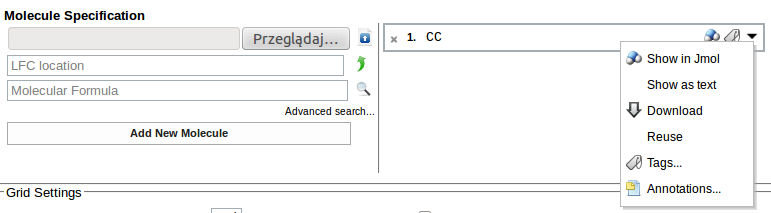
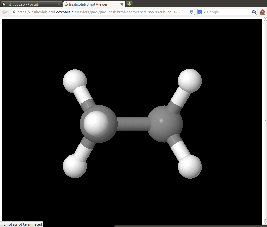
Rozwijając listę menu w oknie zawierającym spis wprowadzonych danych możemy np. obejrzeć na ekranie strukturę wybranego układu wyświetlając ją jako applet Jmol:
Po wybraniu z listy jednego z dostępnych programów wprowadzamy specyfikację wybranej metody i parametrów obliczeń. Rodzaj wprowadzanych danych zależy od wybranego pakietu kwantowochemicznego.
Dla programu Gaussian będzie to krótki opis zadania (Title), parametry wykonania programu określane w sekcji Link 0 (np. limit pamięci), metodę i rodzaj obliczeń w sekcji Route (w przykładzie jest to optymalizacja geometrii z wykorzystaniem funkcjonału B3LYP w bazie 6-31+G*) oraz ładunek i multipletowość badanego układu. Możemy też określić nazwę pliku wynikowego (domyślnie jest to gaussian.log), zażądać zachowania plików "checkpoint" lub wygenerować plik typu cube).
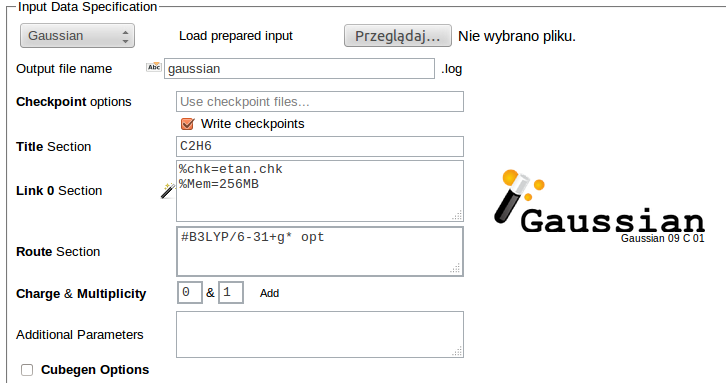
Pomocne przy zadawaniu parametrów w Link 0 section jest użycie ikony obok pola, ułatwiającej poprawny zapis ustawień pamięci i liczby procesorów:
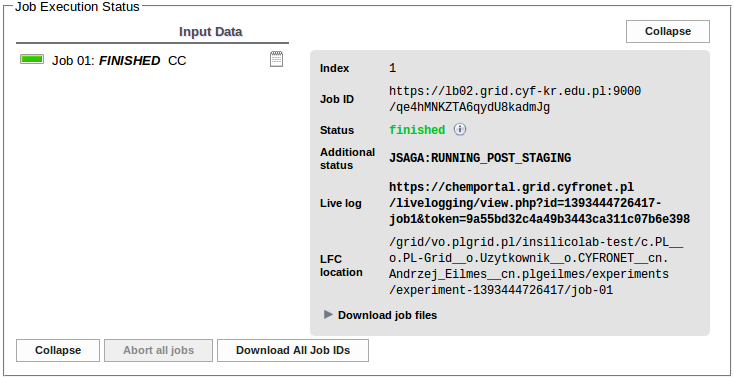
Kliknięciem przycisku Run wydajemy polecenie wysłania zadania do wykonania na infrastrukturze PL-Grid. Aktualny stan zadania możemy śledzić w polu Job Execution Status
Po rozpoczęciu wykonywania obliczeń status ten zmienia się na RUNNING ...
... a po ich zakończeniu na FINISHED.
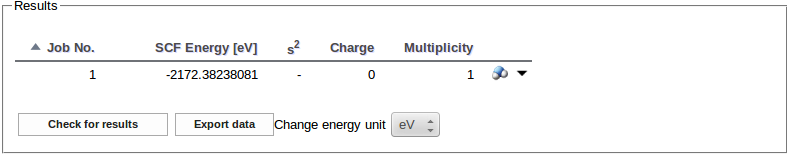
W sekcji Results dostajemy podsumowanie charakterystycznych parametrów wykonanych zadań, np. końcową energię układu po zakończeniu procesu optymalizacji geometrii (możemy wybrać jednostki, w których podawane są wartości energii):
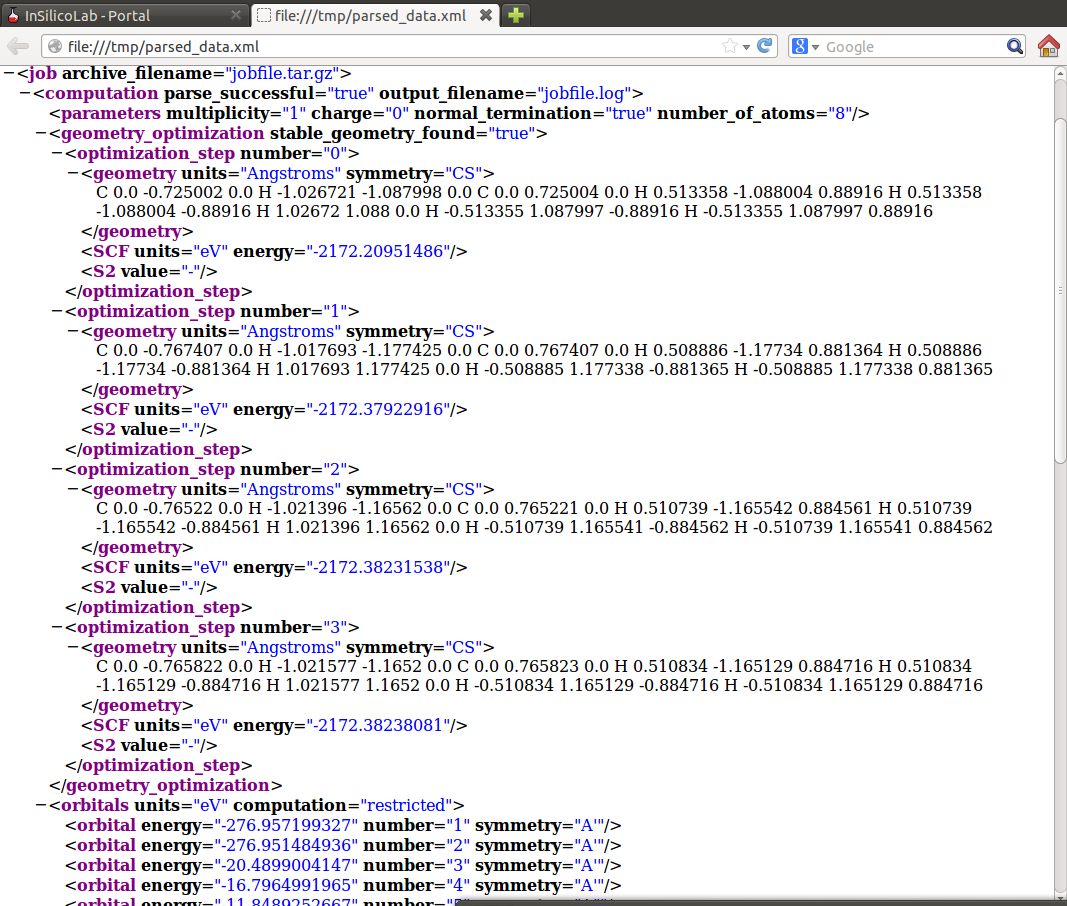
Rozwijając menu Download job files możemy pobrać zarchiwizowane pliki zadania (input, log oraz checkpointy Gaussiana) a także wydobyte z pliku outputowego wyniki obliczeń, np. geometrie i wartości energii w kolejnych krokach optymalizacji geometrii, energie orbitalne i inne parametry (zależnie od rodzaju wykonanych obliczeń).
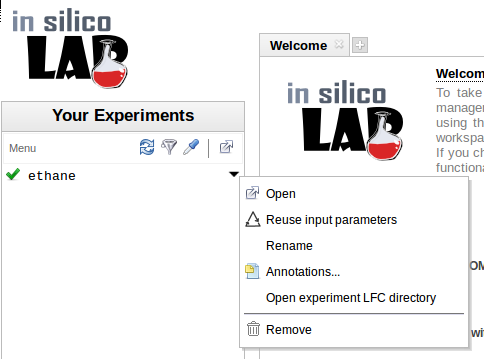
Parametry i dane zakończonych zadań mogą zostać zachowane w spisie eksperymentów użytkownika, w celu późniejszego wykorzystania lub powtórzenia.
Zaawansowane użycie
Po połączeniu się z serwerem usługi
http://insilicolab.chemia.plgrid.pl
...
Pierwszą czynnością jest załadowanie pliku z trajektorią MD, typowo w formacie XYZ. Klikając przycisk Przeglądaj... w menu Load trajectory możemy wybrać żądany plik. Po jego przesłaniu na serwer usługi pojawia się komunikat "Trajectory <filename> loaded" oraz informacja o liczbie ramek znalezionych w trajektorii. W przypadku symulacji dynamiki wykorzystującej periodyczne warunki brzegowe niezbędne jest podanie informacji o długości periodów identyczności po zaznaczeniu opcji Use Periodic Boundary Conditions i wypełnieniu pól z periodami. Jeśli wszystkie trzy wartości są jednakowe, po zaznaczeniu opcji Equal period values wystarczy zadać tylko period A, pozostałe dwie wartości zostaną automatycznie powielone.
...
Wykonywanie obliczeń z użyciem pakietu TeraChem wymaga także aktywowania dodatkowych usług:
- Dostęp do UI - Cyfronet (w dziale "Usługi dostępowe")
- Dostęp do GPGPU - Cyfronet (w dziale "Usługi dostępowe")
- PLG-Data: mini-usługa do zarządzania plikami na klastrze (w dziale "Usługi dostępu do danych")
- Rimrock (w dziale "Usługi inne")
Zwracamy również uwagę że, w związku z przejściem na nowy system scratch na maszynie Zeus, dla osób które używały starego systemu scratch, konieczne jest przełączenie na nowy. Sprawdzenia który system jest używany można dokonać wyświetlając zmienną $SCRATCH (echo $SCRATCH) - powinna ona wskazywać/mnt/lustre/scratch2/people/<nazwa_użytkownika>. Przełączenia na nowy system scratch można dokonać w następujący sposób:
- Należy zalogować się przy użyciu ssh na maszynę zeus.cyfronet.pl (nazwa użytkownika i konta jest tożsama z użytkownikiem i kontem w portalu http://portal.plgrid.pl)
- Usunąć plik ~/.zeusoldscratch (
rm ~/.zeusoldscratch) - Zalogować się ponownie
- Sprawdzić czy zmienna $SCRATCH wskazuje właściwą lokalizację (
echo $SCRATCH) -/mnt/lustre/scratch2/people/<nazwa_użytkownika>
Informacje dotyczące usług i ich aktywowania można znaleźć w rozdziale "Usługi" niniejszego podręcznika.
Pierwsze kroki
Należy połączyć się z serwerem usługi
http://insilicolab.chemia.plgrid.pl/
Po wejściu do usługi, otwiera się ekran. Jeśli nie byliśmy wcześniej zalogowani do usługi, dostępny jest dla nas jedynie ekran anonimowego użytkownika. Logowanie do własnego konta następuje po przyciśnięciu przycisku Log in... i jest możliwe jest na dwa sposoby:
- Logowanie poprzez certyfikat zarejestrowany w przeglądarce użytkownika (nasze dane zostaną sczytane z certyfikatu)
- Logowanie poprzez OpenID dla infrastruktury PL-Grid. W tym wypadku zostaniemy przekierowani na stronę OpenID dla PL-Grid i poproszeni o wpisanie danych dostępowych do infrastruktury - nazwy użytkownika i hasła (tych samych które podajemy w Portalu PL-Grid) lub sczytany zostanie certyfikat umieszczony w przeglądarce. Jeśli usługa InSilicoLab for Chemistry nie była aktywna wcześniej zostanie ona aktywowana automatycznie po zaakceptowaniu odpowiedniego komunikatu. Po zalogowaniu zostaniemy automatycznie przekierowani z powrotem na stronę usługi.
W celu przeprowadzenia obliczeń niezbędne będzie skonfigurowanie odpowiedniego certyfikatu proxy przez kliknięcie na link Configure your proxy:
Certyfikat proxy konieczny jest do przeprowadzania obliczeń z wykorzystaniem infrastruktury PL-Grid a także przeglądania danych zgromadzonych w zasobach tej infrastruktury. Nie jest natomiast konieczny do przeglądania konta użytkownika i zapisanych przez niego analiz a także do wstępnego przygotowania danych przy pomocy narzędzia Trajectory Sculptor (patrz rozdział "Zaawansowane użycie").
Istnieją dwie możliwości konfiguracji proxy w portalu:
- Załadowanie istniejącego proxy - wygenerowanego uprzednio na infrastrukturze. Instrukcja jak wygenerować takie proxy dostępna jest po kliknięciu w pytajnik w polu Upload your proxy. Jest to opcja bezpieczniejsza dla użytkownika, ale wymaga każdorazowego ładowania proxy po upływie jego ważności (12 godzin).
- Generacja proxy z poziomu portalu. Aby wygenerować proxy w portalu, należy załadować do niego pliki certyfikatu i klucza w formacie PEM (najczęściej są to usercert.pem i userkey.pem) lub PKCS12 (np. usercert.p12). Jest to czynność jednorazowa, i nie będzie potrzeby ponawiania jej w przyszłości jeśli certyfikat użytkownika nie ulegnie zmianie. Następnie należy wybrać wirtualną organizację - dla infrastruktury PL-Grid jest to vo.plgrid.pl, podać hasło do załadowanego klucza (UWAGA: hasło nie jest przechowywane nigdzie w systemie) oraz kliknąć przycisk Generate proxy. Zaznaczenie opcji Use MyProxy da nam możliwość automatycznego przedłużania certyfikatu proxy dla uruchomionych zadań do 168. Dodatkowe instrukcje (m. in. instrukcje postępowania w przypadku kiedy certyfikat użytkownika jest w formacie innym niż PEM) dostępne są po kliknięciu w pytajnik w polu VOMS proxy initialization.
W obu przypadkach, jeśli proxy zostało skonfigurowane poprawnie, zostanie wyświetlona na zielono ilość pozostałego czasu ważności proxy w portalu (w polu Your proxy expires in).
Po skonfigurowaniu certyfikatu proxy można przystąpić do właściwego eksperymentu. W celu wykonania obliczeń kwantowochemicznych z menu Create a new experiment należy wybrać pozycję Quantum Chemistry experiment.
W przeglądarce otworzy się karta nowego eksperymentu obliczeniowego wykorzystującego programu chemii kwantowej.
Uwaga: przy wyborze odpowiedniego programu chemii kwantowej, a także przy uzupełnianiu jego parametrów, można skorzystać z usługi Quantum Chemistry Advisor, agregującej dane o tych programach i wspomagającej wybór odpowiedniego do danych obliczeń.
Identyfikację eksperymentu wprowadzamy podając jego krótką nazwę oraz (opcjonalnie) dłuższy opis. W przypadku niepodania krótkiej nazwy, wykorzystane zostanie pole Title formatki aplikacji kwantowochemicznej. Jeśli oba pola zostaną puste, jako krótka nazwa użyty zostanie tekst "(no title)".
Prostą metodą zadania geometrii badanego układu jest wprowadzenie jej przez załadowanie przygotowanego wcześniej pliku w Geometry Specification a następnie określenie jego typu (np. XYZ) w otwierającym się oknie menu. Domyślnie typ pliku ustawiany jest na podstawie jego rozszerzenia. Przyciskiem Finish kończymy wprowadzanie geometrii układu.
Rozwijając listę menu w oknie zawierającym spis wprowadzonych danych możemy np. obejrzeć na ekranie strukturę wybranego układu wyświetlając ją jako applet Jmol:
Po wybraniu z listy jednego z dostępnych programów wprowadzamy specyfikację wybranej metody i parametrów obliczeń. Rodzaj wprowadzanych danych zależy od wybranego pakietu kwantowochemicznego.
Dla programu Gaussian będzie to krótki opis zadania (Title), parametry wykonania programu określane w sekcji Link 0 (np. limit pamięci), metodę i rodzaj obliczeń w sekcji Route (w przykładzie jest to optymalizacja geometrii z wykorzystaniem funkcjonału B3LYP w bazie 6-31+G*) oraz ładunek i multipletowość badanego układu. Możemy też określić nazwę pliku wynikowego (domyślnie jest to gaussian.log), zażądać zachowania plików "checkpoint" lub wygenerować plik typu cube).
Pomocne przy zadawaniu parametrów w Link 0 section jest użycie ikony obok pola, ułatwiającej poprawny zapis ustawień pamięci i liczby procesorów:
Kliknięciem przycisku Run wydajemy polecenie wysłania zadania do wykonania na infrastrukturze PL-Grid. Aktualny stan zadania możemy śledzić w polu Job Execution Status
Po rozpoczęciu wykonywania obliczeń status ten zmienia się na RUNNING ...
... a po ich zakończeniu na FINISHED.
W sekcji Results dostajemy podsumowanie charakterystycznych parametrów wykonanych zadań, np. końcową energię układu po zakończeniu procesu optymalizacji geometrii (możemy wybrać jednostki, w których podawane są wartości energii):
Rozwijając menu Download job files możemy pobrać zarchiwizowane pliki zadania (input, log oraz checkpointy Gaussiana) a także wydobyte z pliku outputowego wyniki obliczeń, np. geometrie i wartości energii w kolejnych krokach optymalizacji geometrii, energie orbitalne i inne parametry (zależnie od rodzaju wykonanych obliczeń).
Parametry i dane zakończonych zadań mogą zostać zachowane w spisie eksperymentów użytkownika, w celu późniejszego wykorzystania lub powtórzenia.
Zaawansowane użycie
Po połączeniu się z serwerem usługi
http://insilicolab.chemia.plgrid.pl
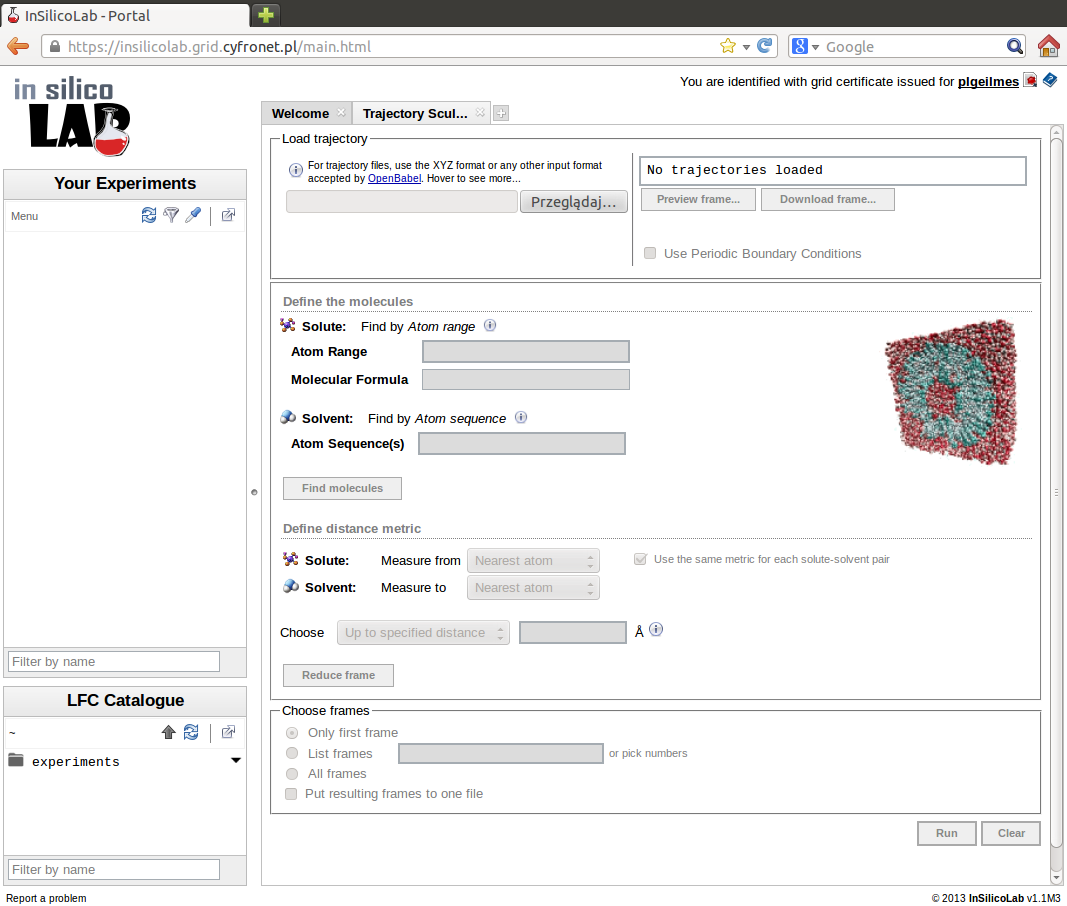
przejściu do portalu InSilicoLab i skonfigurowaniu proxy (patrz Pierwsze kroki) z menu Create a new experiment wybieramy pozycję Trajectory Sculptor. W przeglądarce otworzy się karta eksperymentu pozwalającego przygotować dane do obliczeń kwantowochemicznych poprzez wycięcie wybranych fragmentów układu z ramek trajektorii dynamiki molekularnej.
Pierwszą czynnością jest załadowanie pliku z trajektorią MD, typowo w formacie XYZ. Klikając przycisk Przeglądaj... w menu Load trajectory możemy wybrać żądany plik. Po jego przesłaniu na serwer usługi pojawia się komunikat "Trajectory <filename> loaded" oraz informacja o liczbie ramek znalezionych w trajektorii. W przypadku symulacji dynamiki wykorzystującej periodyczne warunki brzegowe niezbędne jest podanie informacji o długości periodów identyczności po zaznaczeniu opcji Use Periodic Boundary Conditions i wypełnieniu pól z periodami. Jeśli wszystkie trzy wartości są jednakowe, po zaznaczeniu opcji Equal period values wystarczy zadać tylko period A, pozostałe dwie wartości zostaną automatycznie powielone.
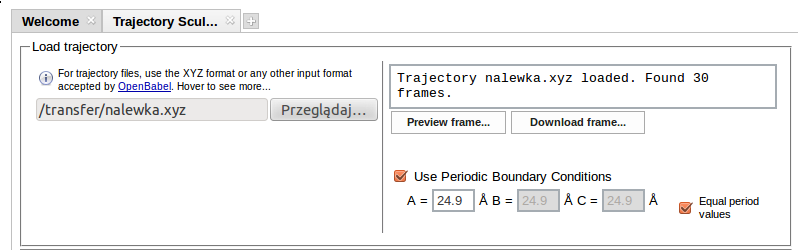
Kolejnym krokiem jest zadanie informacji o cząsteczce rozpuszczonej (jednej w układzie) oraz o cząsteczkach rozpuszczalników. W prezentowanym przykładzie cząsteczką rozpuszczoną była fenyloalanina w roztworze mieszaniny wody i etanolu. Cząsteczkę rozpuszczoną (Solute) podaje się poprzez zakres odpowiadających jej numerów atomów (Atom Range)w ramkach trajektorii, np. 1501-1523. Cząsteczki rozpuszczalnika (Solvent) definiowane są przez podanie ciągu syboli chemicznych (Atom Sequence(s)) tworzących je atomów; sybole te muszą być podane dokładnie w kolejności, w jakiej występują w ramkach trajektorii. Jeśli w układzie występuje więcej niż jeden rodzaj cząsteczek rozpuszczalnika, przyciskiem Add... dodaje się pola dla kolejnych rozpuszczalników. W przykładzie sekwencje symboli opisujących rozpuszczalniki to OHH (woda) oraz OHHCHCHHH (etanol). Po kliknięciu w przycisk Find molecules zanalizowana zostaje pierwsza ramka trajektorii a na ekranie zostają wyświetlone liczby i wzory sumaryczne znalezionych w niej cząsteczek.
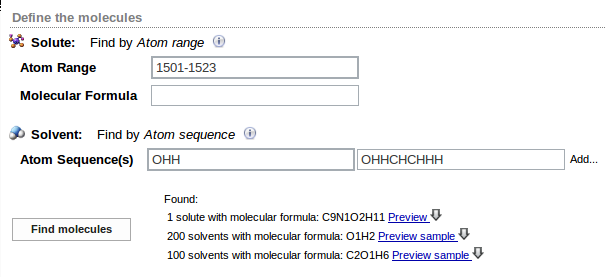
Kliknięcie opcji Preview pozwala obejrzeć model wybranej cząsteczki (np. cząsteczki rozpuszczonej) przy pomocy skryptu JMola:
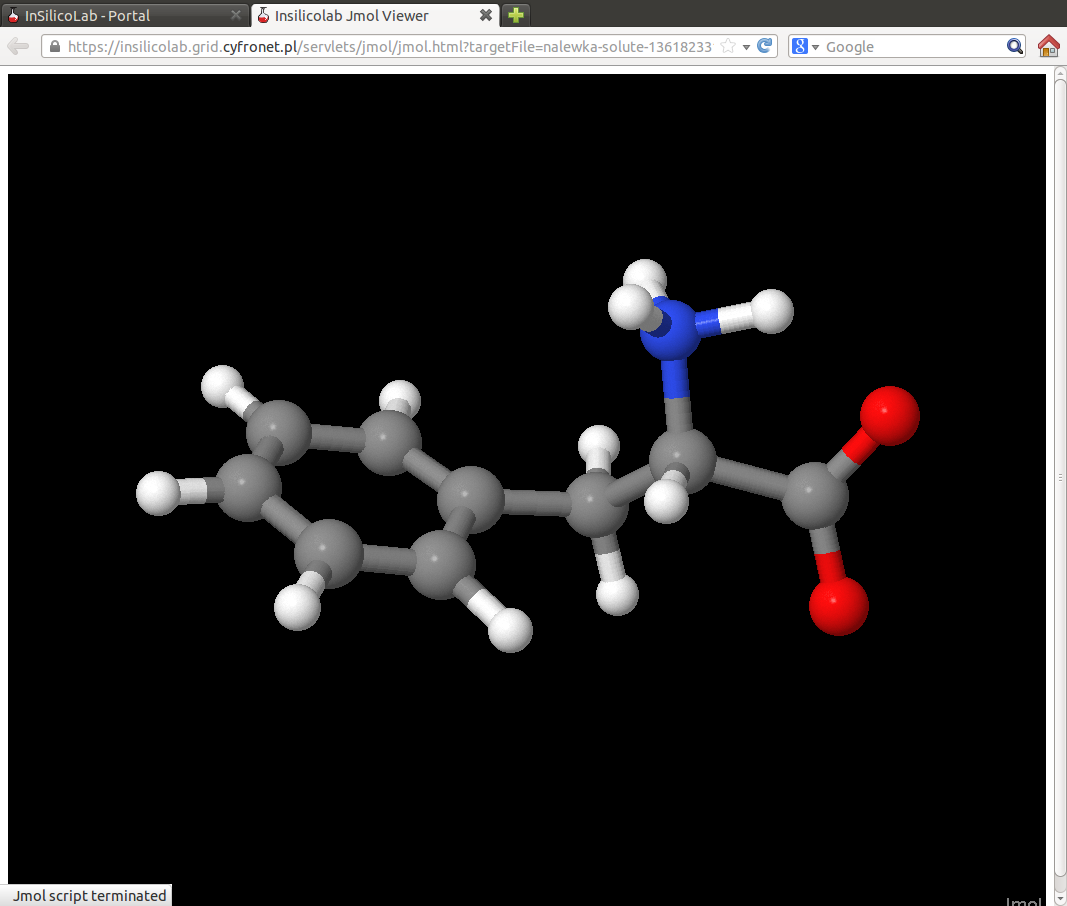
W celu wycięcia z układu cząsteczki rozpuszczonej wraz z otaczającymi ją molekułami rozpuszczalnika niezbędne jest określenie sposobu mierzenia odległości cząsteczka rozpuszczona - rozpuszczalnik (Define distance metric). Najprostszym sposobem jest wybranie Nearest atom zarówno dla Solvent: Measure from jak i Solute: Measure to. Wybór ten oznacza, że odległość między dwiema cząsteczkami jest zdefiniowana jako odległość między najbliższymi sobie atomami obu cząsteczek. Jeśli dodatkowo zaznaczona będzie opcja Use the same metric for each solute-solvent pair, w ten sam sposób mierzona będzie odległość do każdego typu rozpuszczalnika. Następnie określić należy, ile cząstezek rozpuszczalników należy wyciąć z każdej ramki. Opcja Closest molecules oznacza, że wycięta będzie podana liczba molekuł rozpuszczalnika najbliższych (w sensie zdefiniowanej uprzednio metryki) do cząsteczki rozpuszczonej; jeśli liczby te są różne dla różnych typów rozpuszczalników, wartości oddziela się średnikami. W poniższym przykładzie zażądano wycinania 10 cząsteczek wody i 5 cząsteczek etanolu. Kliknięcie przycisku Reduce frame uruchamia wycięcie cząsteczek z przykładowej pierwszej ramki i wyświetlenie liczby wybranych molekuł.
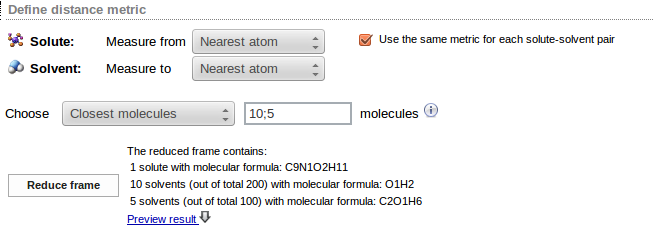
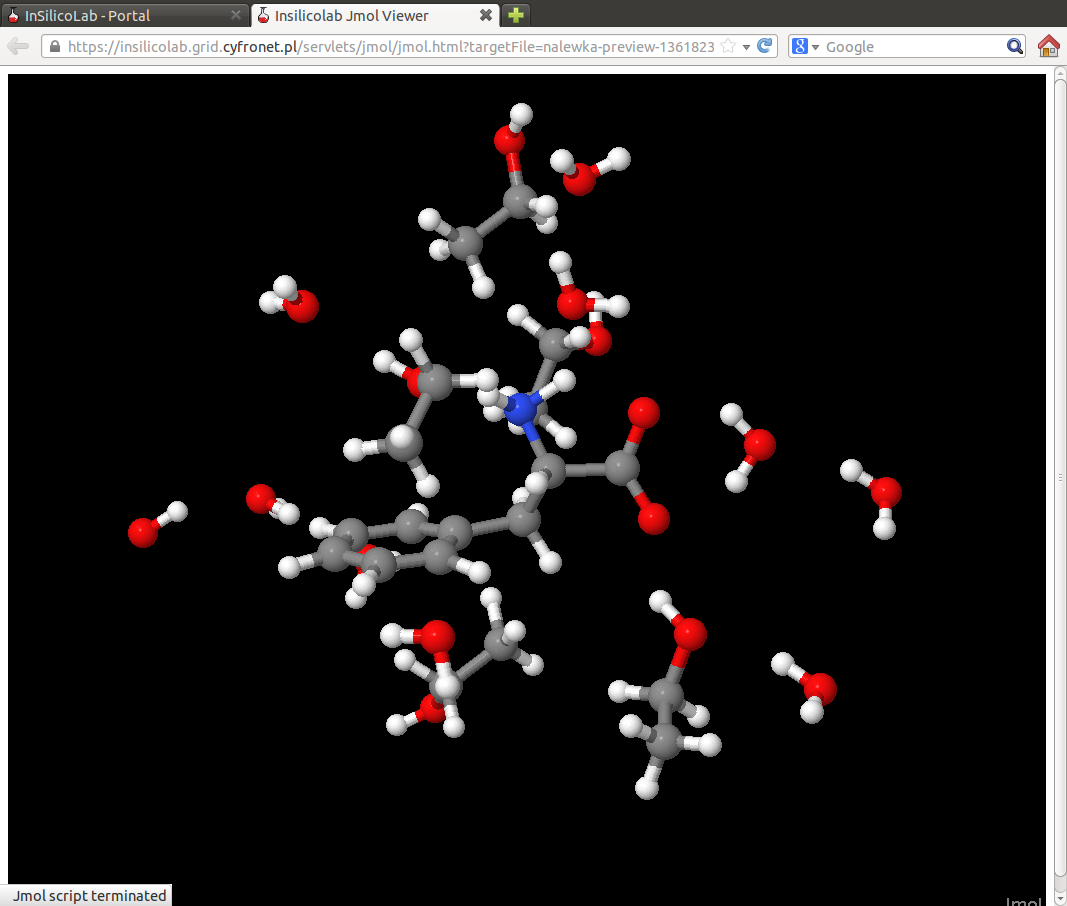
Klikając polecenie Preview result można efekt przycięcia pierwszej ramki obejrzeć w skrypcie JMola, co pozwala użytkownikowi ewentualnie zmodyfikować swój wybór przed przejściem do analizy całej trajektorii:
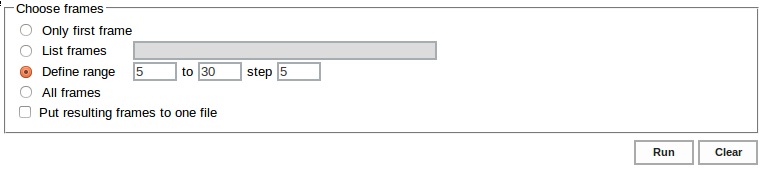
Ostatnim etapem jest wybranie ramek, które mają być analizowane. Możemy wybrać pojedyncze ramki lub zadać zakres i krok wyboru
Dokonawszy wyboru ramek uruchamiamy ich przycinanie za pomocą przycisku Run.
Po zakończeniu analizy trajektorii pojawia się informacja o liczbie utworzonych plików z geometriami przyciętych ramek. Rozwijając menu More możemy przekazać je do eksperymentu kwantowochemicznego (Use all results).
Otwiera się wtedy karta nowego eksperymentu, a w części Geometry Specification widoczna jest lista przekazanych geometrii układów.
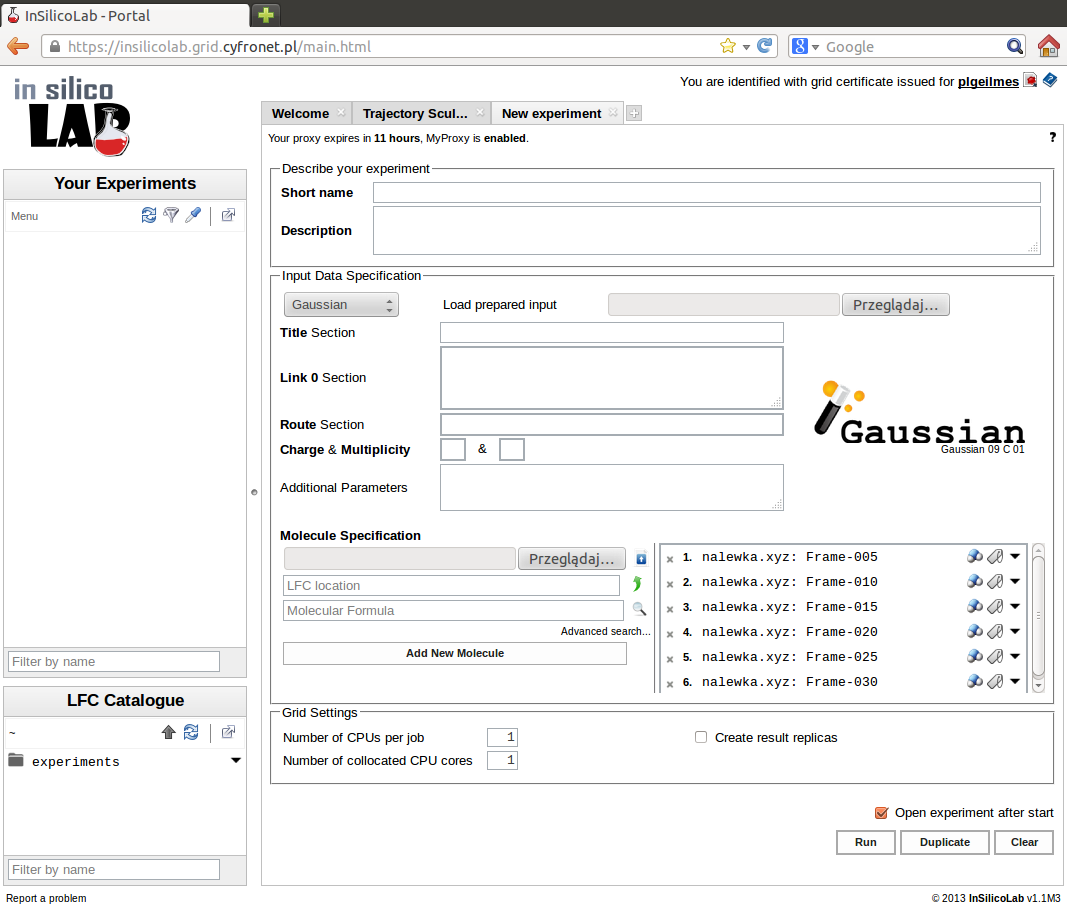
W sposób opisany w sekcji Pierwsze kroki uzupełniamy opis eksperymentu, dokonujemy wyboru programu kwantowochemicznego i zadajemy rodzaj i parametry żądanych obliczeń, np. obliczenie 30 najniższych wzbudzeń elektronowych metodą ZINDO:
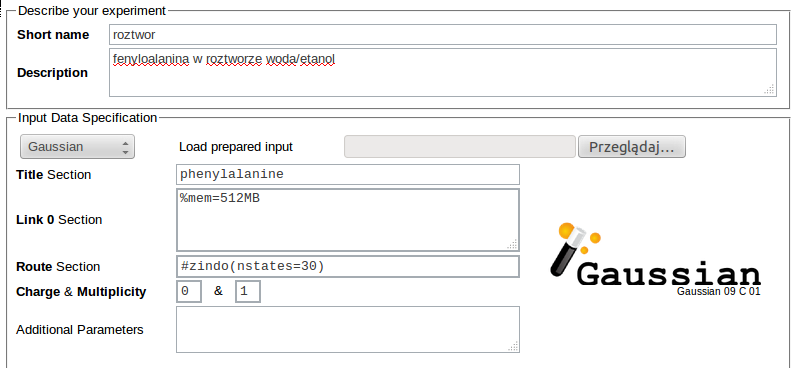
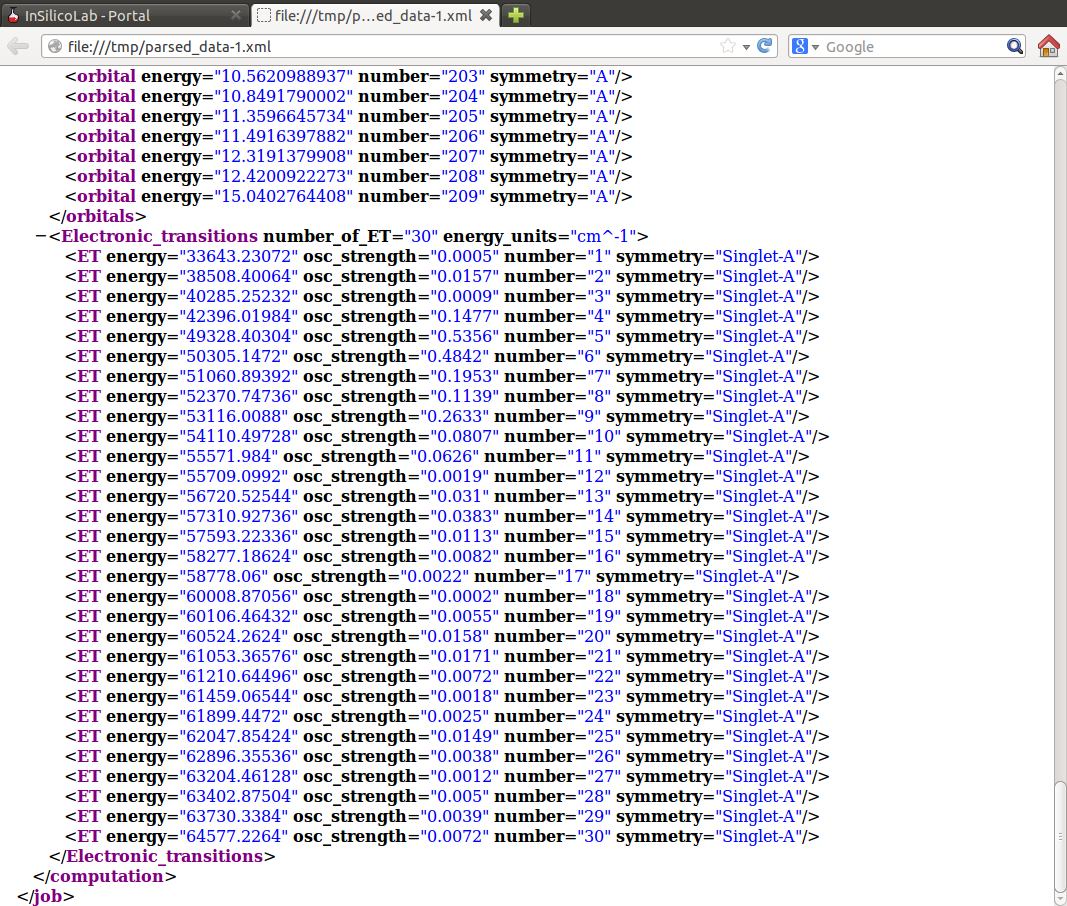
Kliknięcie przycisku Run tworzy pliki wejściowe do programu kwantowochemicznego na podstawie zadanych parametrów oraz geometrii przekazanych z eksperymentu Trajectory Sculptor i wysyła zadania na infrastrukturę gridową.
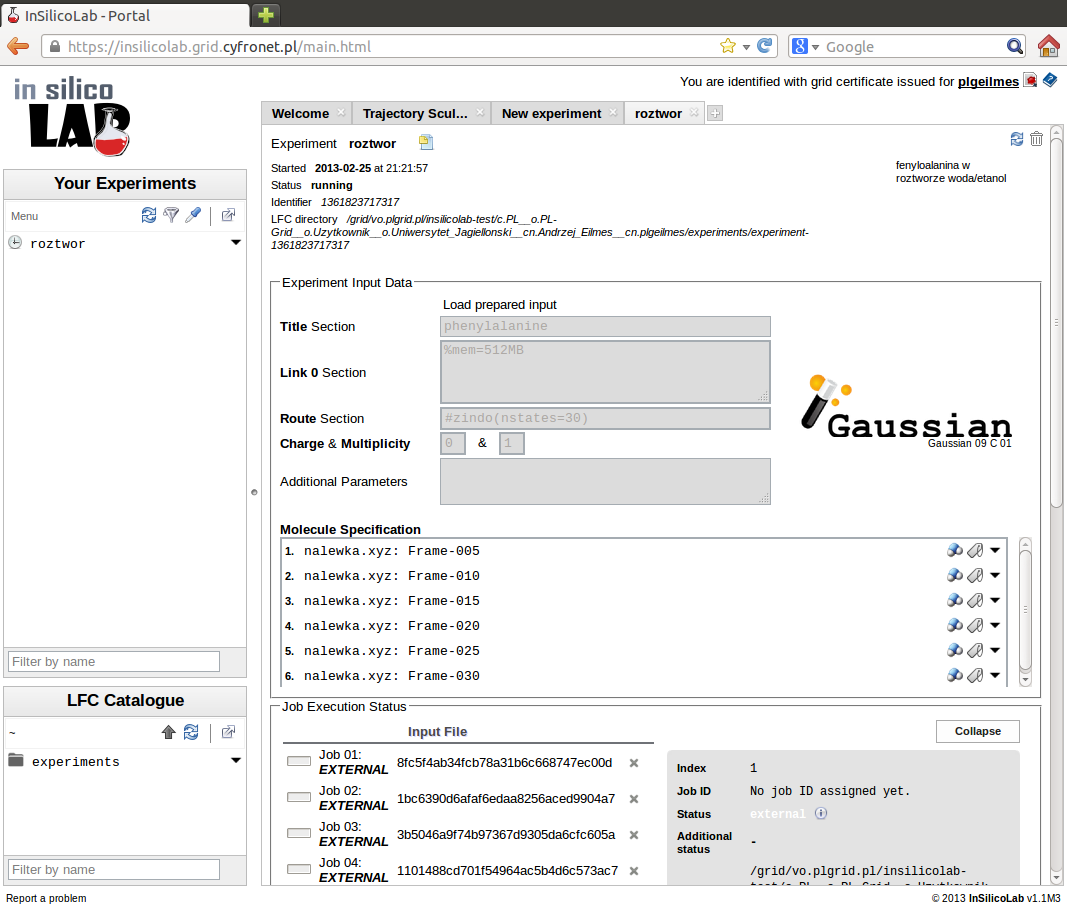
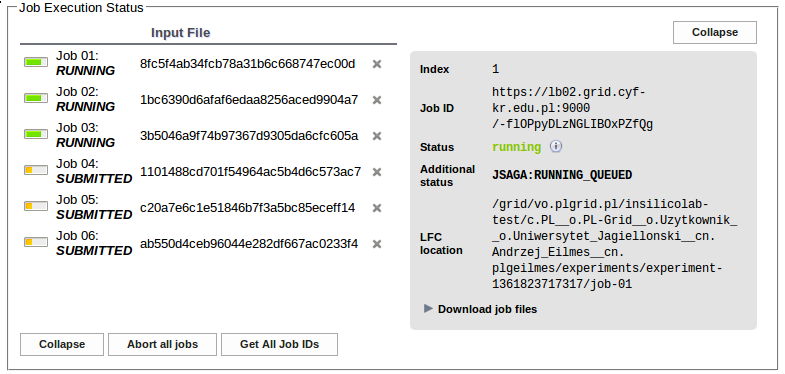
W polu Job Execution Status możemy śledzić jak stan wysłanych zadań zmienia się z EXTERNAL, poprzez SUBMITTED i RUNNING do FINISHED.
W trakcie kończenia się poszczególnych zadań tworzony jest wykres energii SCF oraz tabela z jej wartościami:
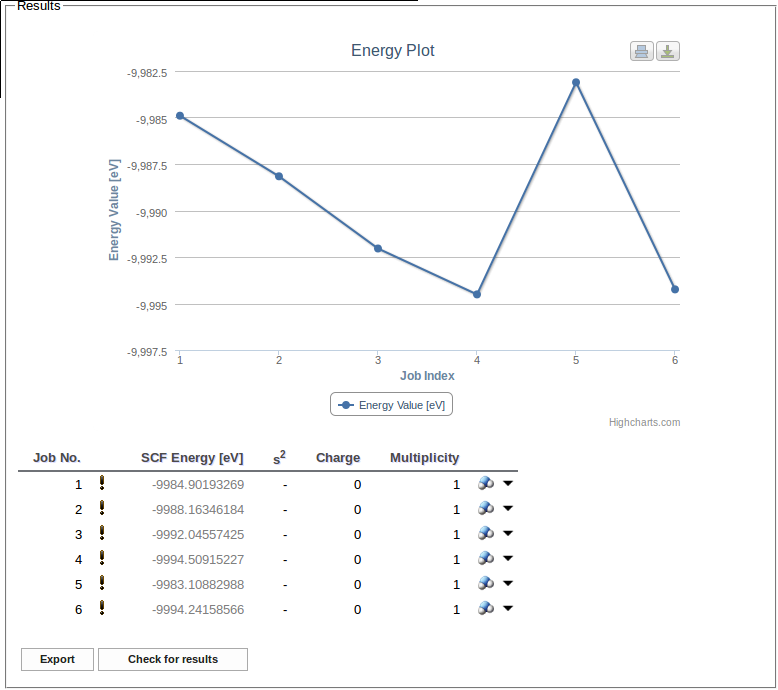
Rozwijając listę Download job files możemy dla wybranego zadania pobrać pliki (dane wejściowe i pliki wynikowe) jako plik .tar.gz oraz plik XML z wybranymi z pliku wynikowego rezultatami obliczeń.
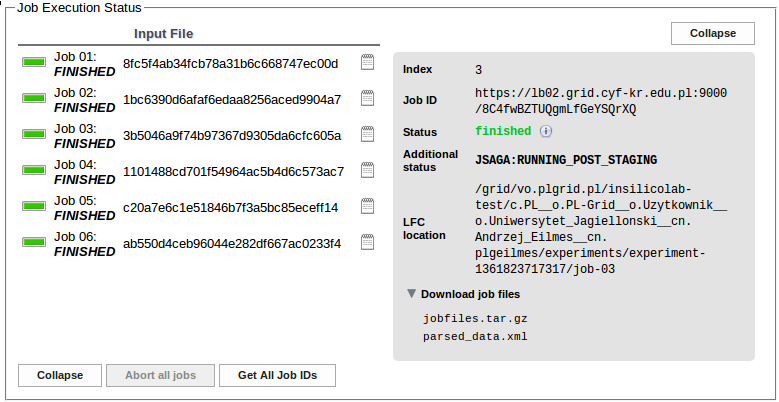
W szczególności dla obliczeń widm elektronowych plik ten zawiera dane o energii przejść oraz ich sile oscylatora:
Użycie programu niedoida w eksperymencie kwantowochemicznym
Program niedoida rozwijany jest na Wydziale Chemii UJ we współpracy z Akademickim Centrum Komputerowym ACK Cyfronet AGH. Implementuje standardowe metody chemii kwantowej (HF, MP2, metody DFT i TD DFT). Unikatowymi cechami programu jest implementacja Laplace-Transform MP2 oraz dressed-TD DFT.
Poniżej przedstawiono użycie programu na przykładzie obliczeń MP2 wykorzystujących transformację Laplace'a przyspieszającą obliczenia dla układów rozciągłych przestrzennie.
Po uruchomieniu z menu ogólnego eksperymentu kwantowochemicznego w sekcji Input Data Specification wybieramy z listy program niedoida.
W sekcji Geometry Specification wprowadzamy współrzędne badanego układu - w przykładzie jest to cząsteczka butadienu. Klikając symbol molekuły możemy zweryfikować poprawność geometrii w applecie JMol.
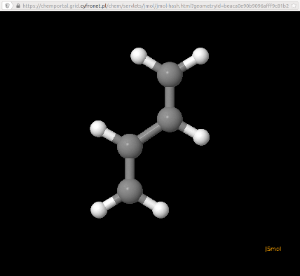
Kolejnym etapem jest uzupełnienie specyfikacji inputu:
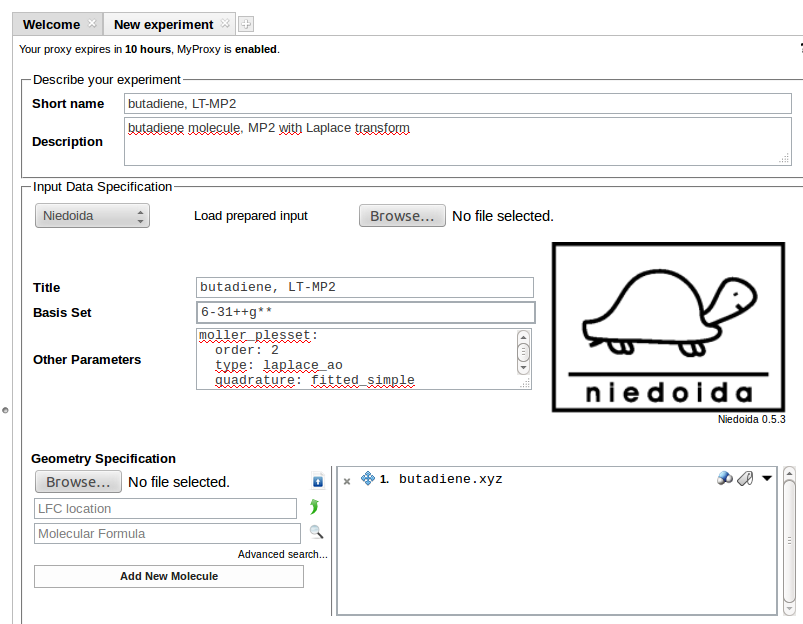
W polu Basis Set podajemy żądaną bazę funkcyjną (w powyższym przykładzie jest to 6-31++G**) a pozostałe elementy specyfikacji obliczeń w polu Other Parameters. W naszym przykładzie jest to:
units: length: angstrom energy: hartreemoller_plesset: order: 2 type: laplace_ao quadrature: fitted_simple
Określiliśmy jednostki odległości i energii oraz zażądaliśmy obliczeń post-HF metodą MP2 z wykorzystaniem transformacji Laplace'a.
Obliczenia uruchamiamy kliknięciem w przycisk Run. Po ich zakończeniu możemy ściągnąć plik outputowy (niedoida.log) oraz plik XML ze sparsowanymi wynikami.
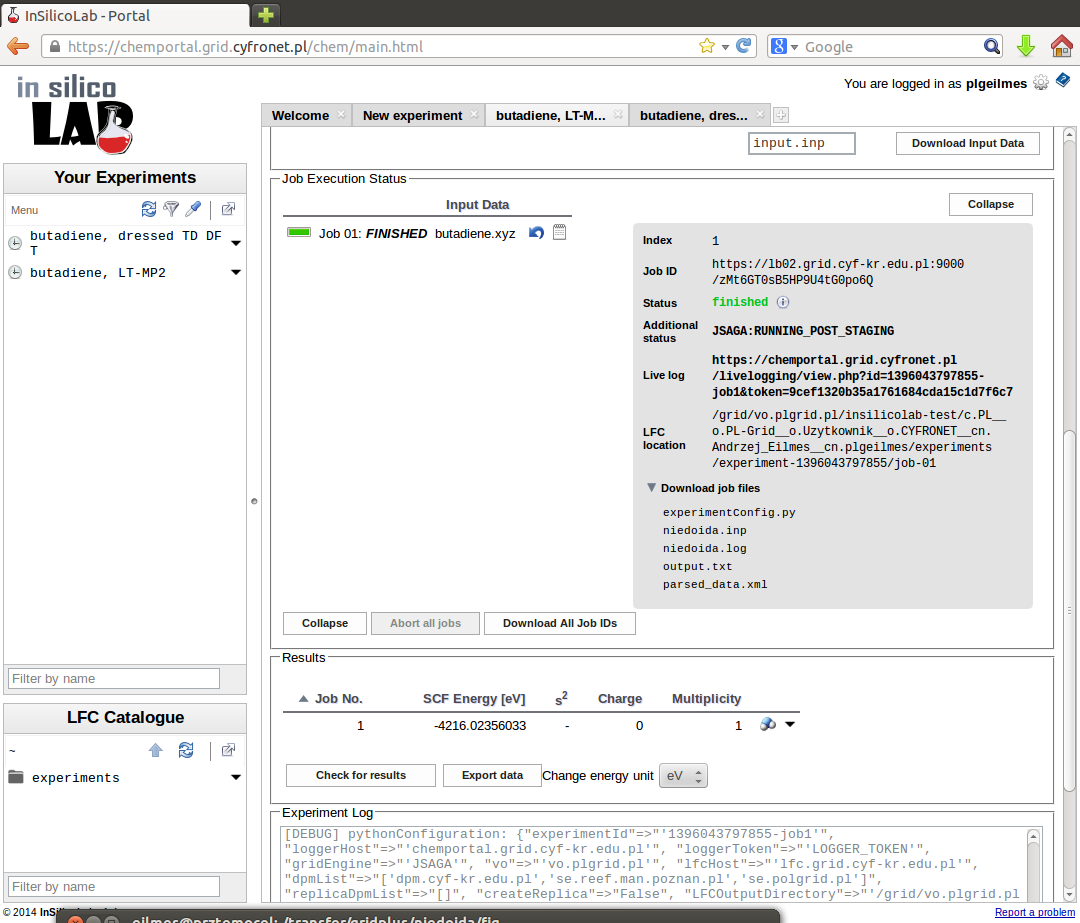
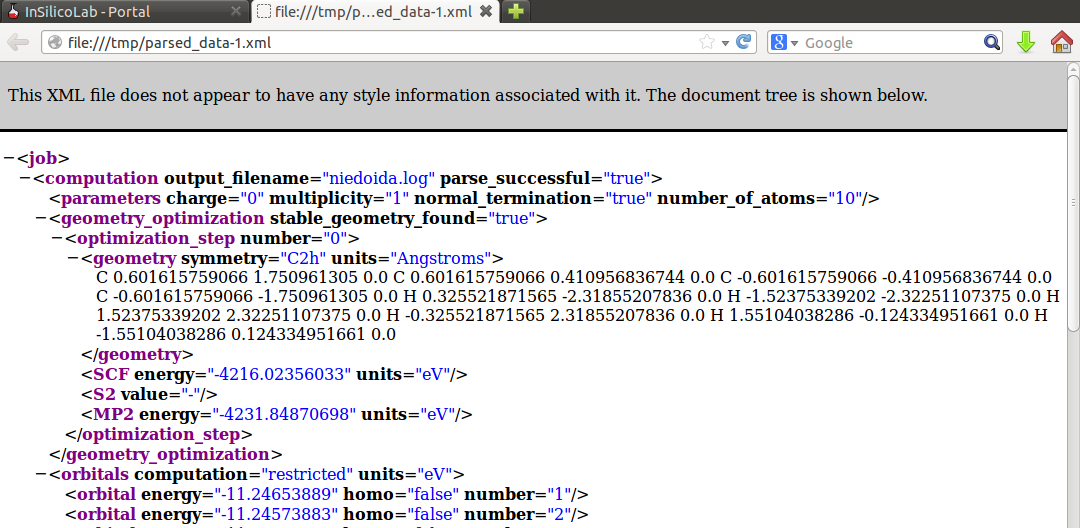
Zwróćmy uwagę, że w polu Results podawana jest energia na poziomie HF SCF, zatem aby odczytać interesującą nas energię w metodzie MP2 należy obejrzeć output programu, lub (wygodniej) odczytać wartość z pliku parsed_data.xml ("MP2 energy"):
Obliczenia kwantowochemiczne na GPU (program TeraChem)
Dostępny na klastrze Zeus program TeraChem pozwala na przyspieszenie części typowych obliczeń kwantowochemicznych poprzez wykonywanie ich na kartach graficznych NVidia.
Uwaga: aby móc skorzystać z obliczeń TeraChemem, należy aktywować dodatkowe usługi - patrz rozdział Aktywowanie usługi.
Szablon odpowiedniego eksperymentu wywołuje się klikając nazwę TeraChem w głównym menu.

Parametry możliwe do ustawienia w eksperymencie to:
- Coordinates file - pozwala na załadowanie pliku w formacie
xyzzawierającego geometrię układu - Basis - baza funkcyjna żądana w obliczeniach
- Charge - całkowity ładunek układu
- Multiplicity - multipletowość układu
- Spin restriction - typ obliczeń restricted lub unrestricted. W przypadku, gdy multipletowość jest większa od 1, wybór typu restricted oznacza uruchomienie obliczeń restricted open-shell.
- Method - metoda kwantowochemiczna. Możliwe jest wykonywanie obliczeń metodą Hartree-Focka lub obliczeń DFT z użyciem różnych funkcjonałów
- Dispersion - pozwala uwzględnić w obliczeniach empiryczną poprawkę dyspersyjną Grimmego (w wersji D2 lub D3)
- Calculation type - rodzaj obliczeń: obliczenia energii (single point), optymalizacja geometrii, poszukiwanie stanu przejściowego, dynamika Borna-Oppenheimera, obliczenie energii i gradientu oraz projekcja funkcji falowej
- QM/MM - zaznaczenie tej opcji pozwala na uwzględnienie wody traktowanej metodą mechaniki molekularnej
- Additional parameters - pole pozwalające na podanie innych słów kluczowych programu
- GPU count - liczba kart graficznych, na których ma zostać uruchomiony program (od 1 do 8)
- Use a non-default grant - standardowo obliczenia wykonywane są w ramach zasobów z domyślnego grantu użytkownika PL Grid. Zaznaczenie tej opcji pozwala na podanie nazwy innego grantu obliczeniowego użytkownika
Jeśli jako Calculation type wybrano Born-Oppenheimer Dynamics, w dodatkowych polach zadaje się:
- Maximum steps - liczbę kroków dynamiki
- Initial temperature - temperaturę określającą początkowy rozkład prędkości
- Thermostat temperature - temperaturę, która ma być utrzymywana w trakcie obliczeń
- Temperature control - sposób utrzymywania zadanej temperatury: termostat Langevina lub skalowanie prędkości
Wybór opcji QM/MM pozwala na wykonanie obliczeń, w których część układu traktowana jest na poziomie wybranej metody kwantowochemicznej a otaczające ją cząsteczki wody opisywane są mechaniką molekularną w parametryzacji TIP3P. W takim przypadku należy użyć listy TIP3P water molecules do załadowania pliku xyz z geometrią cząsteczek wody (część kwantowochemiczna zdefinowana jest przez plik podany w Coordinates file). Cząsteczki wody w pliku zapisywane są kolejno, atomy cząsteczki muszą być podawane w kolejności OHH.
W poniższym przykładzie zażądano wykonania 1000 kroków symulacji dynamiki Borna-Oppenheimera z początkową temperaturą 350 K oraz żądaną temperaturą układu 350 K utrzymywaną przez termostat Langevina. Wybrano kombinowaną metodę QM/MM. Geometria kwantowochemicznej części układu zawarta jest w pliku mc1.xyz, jej ładunek i multipletowość wynoszą 0 i 1. Obliczenia kwantowochemiczne mają zostać wykonane metodą RB3LYP w bazie 6-31G z uwzględnieniem poprawki dyspersyjnej D3. Gęstość gridu używanego w obliczeniach DFT ustawiono na poziomie 2. Kwantowochemiczna część układu otoczona jest cząsteczkami wody TIP3P o początkowej geometrii zadanej w plikumc1-w.xyz. Obliczenia mają być wykonane z użyciem 8 kart GPU w ramach domyślnego grantu użytkownika.

Kliknięcie przycisku Run powoduje przesłanie zadania do wykonania na klastrze Zeus.
Po zakończeniu obliczeń użytkownik ma możliwość pobrania pliku log (output programu TeraChem) oraz pliku tar z zawartością katalogu scr (pozostałe pliki wynikowe tworzone przez program).
Kliknięcie opcji Preview pozwala obejrzeć model wybranej cząsteczki (np. cząsteczki rozpuszczonej) przy pomocy skryptu JMola:
W celu wycięcia z układu cząsteczki rozpuszczonej wraz z otaczającymi ją molekułami rozpuszczalnika niezbędne jest określenie sposobu mierzenia odległości cząsteczka rozpuszczona - rozpuszczalnik (Define distance metric). Najprostszym sposobem jest wybranie Nearest atom zarówno dla Solvent: Measure from jak i Solute: Measure to. Wybór ten oznacza, że odległość między dwiema cząsteczkami jest zdefiniowana jako odległość między najbliższymi sobie atomami obu cząsteczek. Jeśli dodatkowo zaznaczona będzie opcja Use the same metric for each solute-solvent pair, w ten sam sposób mierzona będzie odległość do każdego typu rozpuszczalnika. Następnie określić należy, ile cząstezek rozpuszczalników należy wyciąć z każdej ramki. Opcja Closest molecules oznacza, że wycięta będzie podana liczba molekuł rozpuszczalnika najbliższych (w sensie zdefiniowanej uprzednio metryki) do cząsteczki rozpuszczonej; jeśli liczby te są różne dla różnych typów rozpuszczalników, wartości oddziela się średnikami. W poniższym przykładzie zażądano wycinania 10 cząsteczek wody i 5 cząsteczek etanolu. Kliknięcie przycisku Reduce frame uruchamia wycięcie cząsteczek z przykładowej pierwszej ramki i wyświetlenie liczby wybranych molekuł.
Klikając polecenie Preview result można efekt przycięcia pierwszej ramki obejrzeć w skrypcie JMola, co pozwala użytkownikowi ewentualnie zmodyfikować swój wybór przed przejściem do analizy całej trajektorii:
Ostatnim etapem jest wybranie ramek, które mają być analizowane. Możemy wybrać pojedyncze ramki lub zadać zakres i krok wyboru
Dokonawszy wyboru ramek uruchamiamy ich przycinanie za pomocą przycisku Run.
Po zakończeniu analizy trajektorii pojawia się informacja o liczbie utworzonych plików z geometriami przyciętych ramek. Rozwijając menu More możemy przekazać je do eksperymentu kwantowochemicznego (Use all results).
Otwiera się wtedy karta nowego eksperymentu, a w części Geometry Specification widoczna jest lista przekazanych geometrii układów.
W sposób opisany w sekcji Pierwsze kroki uzupełniamy opis eksperymentu, dokonujemy wyboru programu kwantowochemicznego i zadajemy rodzaj i parametry żądanych obliczeń, np. obliczenie 30 najniższych wzbudzeń elektronowych metodą ZINDO:
Kliknięcie przycisku Run tworzy pliki wejściowe do programu kwantowochemicznego na podstawie zadanych parametrów oraz geometrii przekazanych z eksperymentu Trajectory Sculptor i wysyła zadania na infrastrukturę gridową.
W polu Job Execution Status możemy śledzić jak stan wysłanych zadań zmienia się z EXTERNAL, poprzez SUBMITTED i RUNNING do FINISHED.
W trakcie kończenia się poszczególnych zadań tworzony jest wykres energii SCF oraz tabela z jej wartościami:
Rozwijając listę Download job files możemy dla wybranego zadania pobrać pliki (dane wejściowe i pliki wynikowe) jako plik .tar.gz oraz plik XML z wybranymi z pliku wynikowego rezultatami obliczeń.
W szczególności dla obliczeń widm elektronowych plik ten zawiera dane o energii przejść oraz ich sile oscylatora:
Użycie programu niedoida w eksperymencie kwantowochemicznym
Program niedoida rozwijany jest na Wydziale Chemii UJ we współpracy z Akademickim Centrum Komputerowym ACK Cyfronet AGH. Implementuje standardowe metody chemii kwantowej (HF, MP2, metody DFT i TD DFT). Unikatowymi cechami programu jest implementacja Laplace-Transform MP2 oraz dressed-TD DFT.
Poniżej przedstawiono użycie programu na przykładzie obliczeń MP2 wykorzystujących transformację Laplace'a przyspieszającą obliczenia dla układów rozciągłych przestrzennie.
Po uruchomieniu z menu ogólnego eksperymentu kwantowochemicznego w sekcji Input Data Specification wybieramy z listy program niedoida.
W sekcji Geometry Specification wprowadzamy współrzędne badanego układu - w przykładzie jest to cząsteczka butadienu. Klikając symbol molekuły możemy zweryfikować poprawność geometrii w applecie JMol.
Kolejnym etapem jest uzupełnienie specyfikacji inputu:
W polu Basis Set podajemy żądaną bazę funkcyjną (w powyższym przykładzie jest to 6-31++G**) a pozostałe elementy specyfikacji obliczeń w polu Other Parameters. W naszym przykładzie jest to:
...
Określiliśmy jednostki odległości i energii oraz zażądaliśmy obliczeń post-HF metodą MP2 z wykorzystaniem transformacji Laplace'a.
Obliczenia uruchamiamy kliknięciem w przycisk Run. Po ich zakończeniu możemy ściągnąć plik outputowy (niedoida.log) oraz plik XML ze sparsowanymi wynikami.
Zwróćmy uwagę, że w polu Results podawana jest energia na poziomie HF SCF, zatem aby odczytać interesującą nas energię w metodzie MP2 należy obejrzeć output programu, lub (wygodniej) odczytać wartość z pliku parsed_data.xml ("MP2 energy"):
...
Gdzie szukać dalszych informacji?
...
Overview
Content Tools