Page History
InSilicoLab for Chemistry
InSilicoLab is a collection of tools intended to assist the researcher in performing complex experiments of computational chemistry. It facilitates preparation of input files and submitting the jobs to the grid infrastructure, controls job execution, collects the output files and performs basic analysis of results. The user-defined experiment can be stored to allow for easy repetition of computational procedure. InSilicoLab integrates with the grid storage providing access to the user data.
...
Note: It is advisable to start reading with the basic user's manual before advancing to this one.
Getting access to InSilicoLab
InSilicoLab is accessible to anyone registered as a PL-Grid user or having a valid Grid Certificate and belonging to vo.plgrid.pl or gaussian Virtual Organisations. PL-Grid users may log in using their PL-Grid credentials by choosing to log in with OpenID or with personal certificate. In case of OpenID login, the user will be redirected to the OpenID page, where they will be prompted for PL-Grid login and password or a Grid Certificate associated with the account. If certificate login is chosen, the InSilicoLab portal will read the certificate installed in the user's browser. Note: If the certificate is not present or the user declines to use it for authentication, browser restart would be required to log in with the certificate again.
...
InSilicoLab offers also anonymous preview access. It includes manipulation of molecular dynamics trajectories with use of Trajectory Sculptor and viewing sample experiments and their data. Downloading any data is not possible in the anonymous mode.
Trajectory Sculptor
Sequential MD/QC approach is often used in explicit solvent modeling. In the first step, Molecular Dynamics simulations (classical or ab initio) are employed to generate atomistic structures of the system and their evolution in time. In the next step, quantum-chemical calculations are performed for selected part of the system (solute molecule and closest solvent molecules in its solvation shell) to calculate required properties (such as potential energy, excitation energies, chemical shifts, etc.). Such calculations are usually repeated for series of frames chosen from the trajectory.
...
All steps of MD trajectory analysis in Trajectory Sculptor are described in detail in following sections.
Accessing Trajectory Sculptor
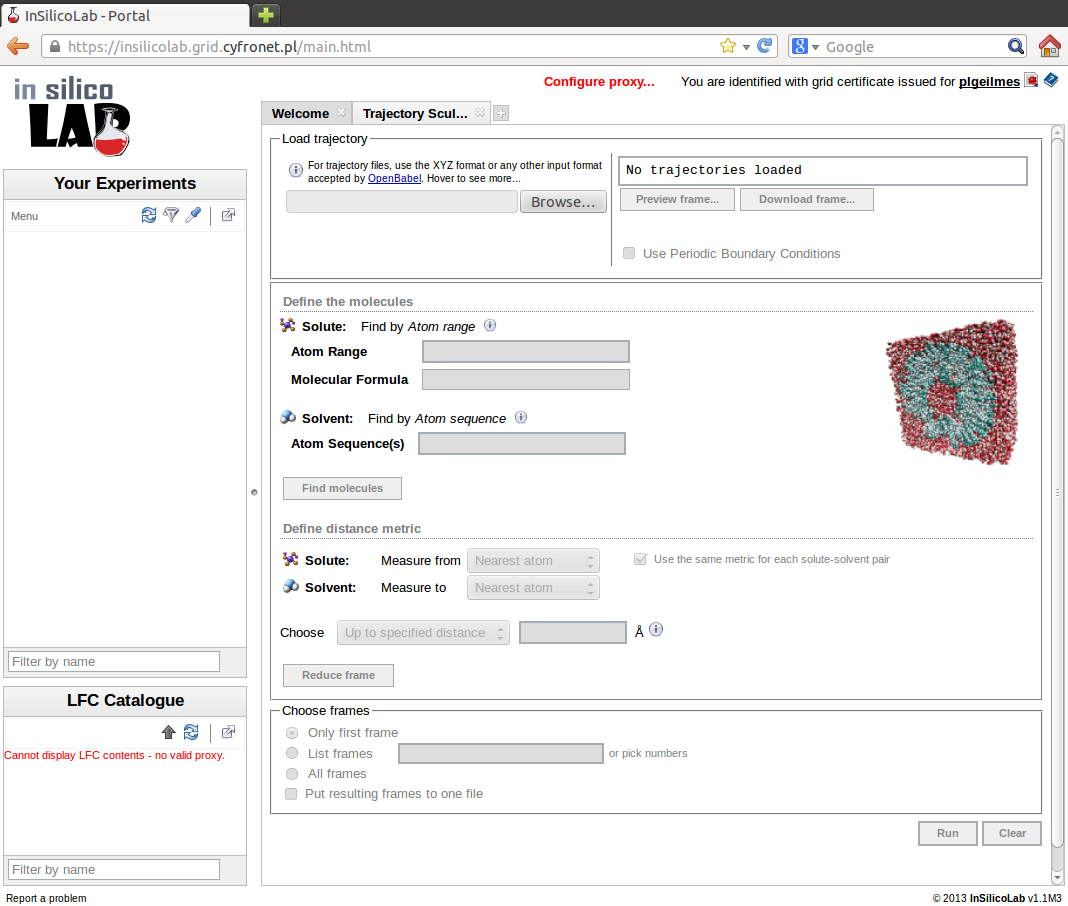
Trajectory Sculptor can be accessed from the main page after entering the portal. After choosing Trajectory Sculptor from the list of new experiments to create, the Trajectory Sculptor interface, similar to the one in the picture is shown.
Trajectory upload
Basic format of the MD trajectory files used by Trajectory Sculptor is the xyz format. However, trajectory may be supplied in other format recognized by OpenBabel in which case it will be automatically converted to xyz file.
...
PBC information for the system is stored within the Trajectory Sculptor. If the user downloads the same file in the future, PBC settings can be easily recalled using the Insert saved settings option.
System specification
The next step of MD trajectory analysis is the specification of molecules (solute and solvent) present in the system. Specification is entered in the Define the molecules panel.
...
In the above example one solute molecule consisting of 23 atoms with the formula C9NO2H11, 200 water molecules (defined as OHH sequence) and 100 ethanol molecules (defined as OHHCHCHHH) are found in the frame. Solute molecule and example of solvent molecules may be viewed on choosing Preview or Preview sample option, respectively. Their geometries may be downloaded in the xyz format on pressing the arrow symbol.
Distance metric definition
To select solvent molecules closest to the solute one must define how the distance between two molecules is defined. Trajectory Sculptor allows several ways of measuring solute-solvent distances.
...
Checking the Use the same metric for each solute-solvent pair option facilitates entering the same definition for all solvents. Note, however, that this option is inactive if the Chosen atom reference point has been selected for the solute.
Selection of the solvation shell
After the solute-solvent distance metric has been defined it is possible to specify which solvent molecules should be included in the solvation shell. This is achieved by proper settings of the values in Choose option. Solvent molecules can be specified in two ways:
...
Distance metric definition and the parameters of the Choose option may be changed as many times as necessary, therefore the Preview result command helps to find out the best distance metric and the appropriate way of selecting solvent molecules.
Final analysis of the trajectory
When the molecules in the system have been defined, the distance metric and the way of selecting molecules have been set up, the MD trajectory can be finally analyzed to extract solute with specified solvation shell from selected frames.
...
They can be downloaded individually (Download), downloaded as one xyz file (Download all) stored or passed to another InSilicoLab experiment (Use all results).
Quantum-chemical calculations experiment
This experiment facilitates preparation of input files for quantum-chemical calculations and execution of jobs on the PLGrid infrastructure.
Accessing the QC experiment
QC experiment can be accessed from the main page after entering the portal - by choosing Quantum Chemistry experiment from the list of new experiments to create.
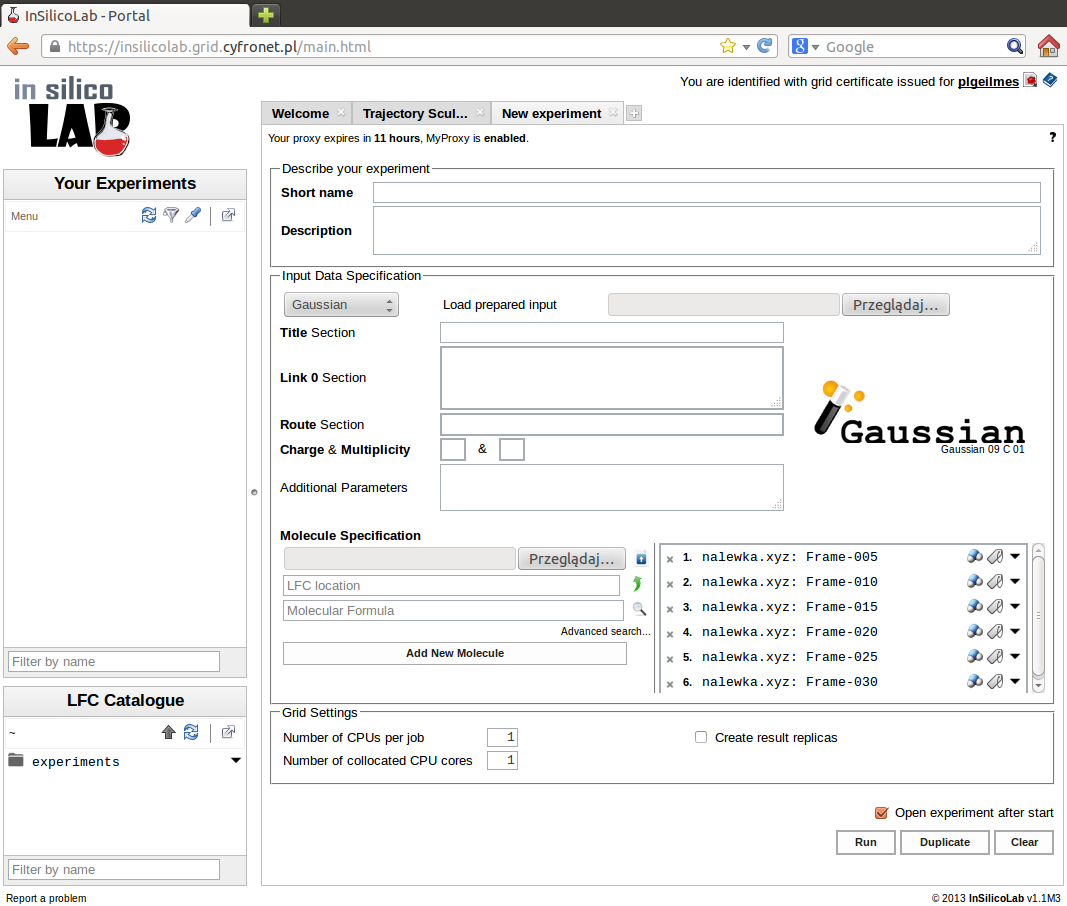
Another possibility is to invoke the QC experiment from another InSilicoLab experiment (e.g. Trajectory Sculptor). In such case geometries of the system may be passed between experiments and they are immediately available as input data in the QC experiment interface.
Setting-up the QC experiment
The QC experiment is identified by the name entered in the field Short name. Optionally, more detailed description may be provided in the Description field. If no Short name is supplied, the contents of the Title section will be used. If both fields are empty, the "(no title)" will be used as a short name.
...
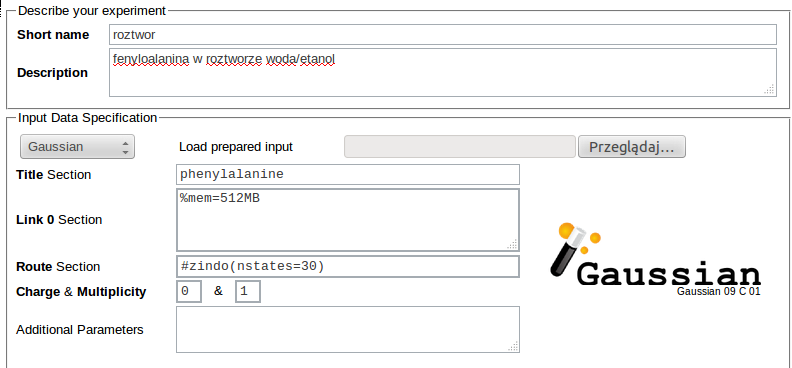
For example, the input for Gaussian ZINDO calculations of 30 lowest electronic excitations for a phenylalanine molecule in a solution using structures prepared in Trajectory Sculptor and requesting 512MB RAM for each job may read:
Job execution
Clicking the Run button creates the input files based on the supplied geometries and parameters entered in the Input Data Specification. The jobs are then submitted for execution to the PLGrid.
...
User can store the parameters of the experiment and job files for future retrieval or easy repetition of calculations:
Using niedoida program in general quantum-chemistry experiment
Niedoida is a quantum-chemical package developed at the Faculty of Chemistry of the Jagiellonian University in collaboration with the Academic Computer Centre ACK Cyfronet AGH.
...
Program features implementation of "dressed"-TD DFT incorporating corrections from doubly-excited states.
LT-MP2 calculations example
To use the program its name needs to be selected from the list of programs in the Input Data Specification section:
...
Note that in the Results window the HF SCF energy is displayed, therefore to read the MP2 energy log file needs to be inspected or (more conveniently) the requested value can be found in the parsed_data.xml file:
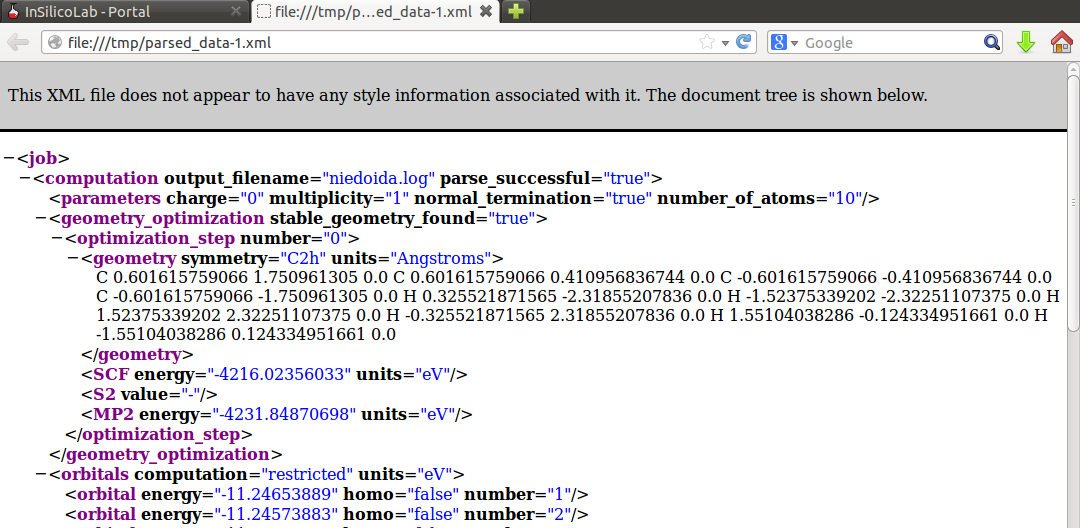
dressed-TD DFT calculations example
Selection of the program and entering the coordinates of butadiene molecule can be done as in the former example.
...
In the above example corrections arising from double excited states lowered the energy of the 5th state from 6.865 to 6.438 eV.
Cubegen
This experiment is used to generate Gaussian cube files.
...
Overview
Content Tools