Page History
Krótki opis usługi
Usługa jest przeznaczona dla biologów oraz bioinformatyków.
Chipster jest implementacją popularnego środowiska zarządzania zadaniami pozwalającego na uproszczone uruchamianie analiz bioinformatycznych na zasobach obliczeniowych PLGrid. W ramach usługi szczególny nacisk położony został na udostępnienie możliwie dużej liczby narzędzi związanych z analizą danych pochodzących z eksperymentów opartych o metody wysokoprzepustowego sekwencjonowania. Wykonywanie analiz z użyciem Chipster oparte jest o wygodny interfejs dostępny w formie aplikacji Java, pozwalający na intuicyjne zarządzanie danymi, narzędziami oraz wynikami. Wbudowane moduły wizualizacji pozwalają na przejrzystą i efektywną analizę wyników.
Aktywowanie usługi
Aby skorzystać z usługi Chipster, należy mieć aktywne konto w infrastrukturze PLGrid.
Do usługi Chipster może uzyskać dostęp każdy użytkownik PLGrid, który jest użytkownikiem usługi Molecular Biology Data Analysis Toolkit. W celu uzyskania dostępu do tych usług należy wejść na stronę http://portal.plgrid.pl, a następnie zalogować się podając swój identyfikator PLGrid (np. plgkowalski) i hasło do portalu, po czym z górnej belki zawierającej menu wybrać opcję „Moje konto”. W prawej kolumnie ukaże się Katalog usług dostępnych dla danego użytkownika.
Z katalogu usług należy wybrać kategorię: Platforma Dziedzinowa – Biologia, a następnie „rozwiń”, ukaże się lista usług w tej kategorii, wśród nich będzie usługa o nazwie Molecular Biology Data Analysis Toolkit a tuż obok link zatytułowany „aplikuj o usługę”. Akceptacja użytkownika następuje automatycznie. Gdy rejestracja się powiedzie, usługa na liście w katalogu zyska status „aktywny”.
Pierwsze kroki
Uruchomienie usługi
Uruchomienie aplikacji Chipster możliwe jest na każdym komputerze wyposażonym w przeglądarkę internetową oraz środowisko Java Web Start. Usługa dostępna jest poprzez portal MBDAT bądź pod adresem: http://chipster.biologia.plgrid.pl. Na wskazanej stronie należy wcisnąć link launch Chipster . Alternatywnie, można również wybrać wersję aplikacji możliwą do zastosowania na komputerach o większej ilości pamięci RAM: 3 lub 6 GB. Po wciśnięciu linku pobrany zostanie plik startowy aplikacji chipster.jnlp . Pobrany plik należy uruchomić za pomocą Java Web Start. Podczas uruchamiania aplikacji nastąpi monit o zalogowanie z użyciem danych konta PLGrid.
Organizacja interfejsu
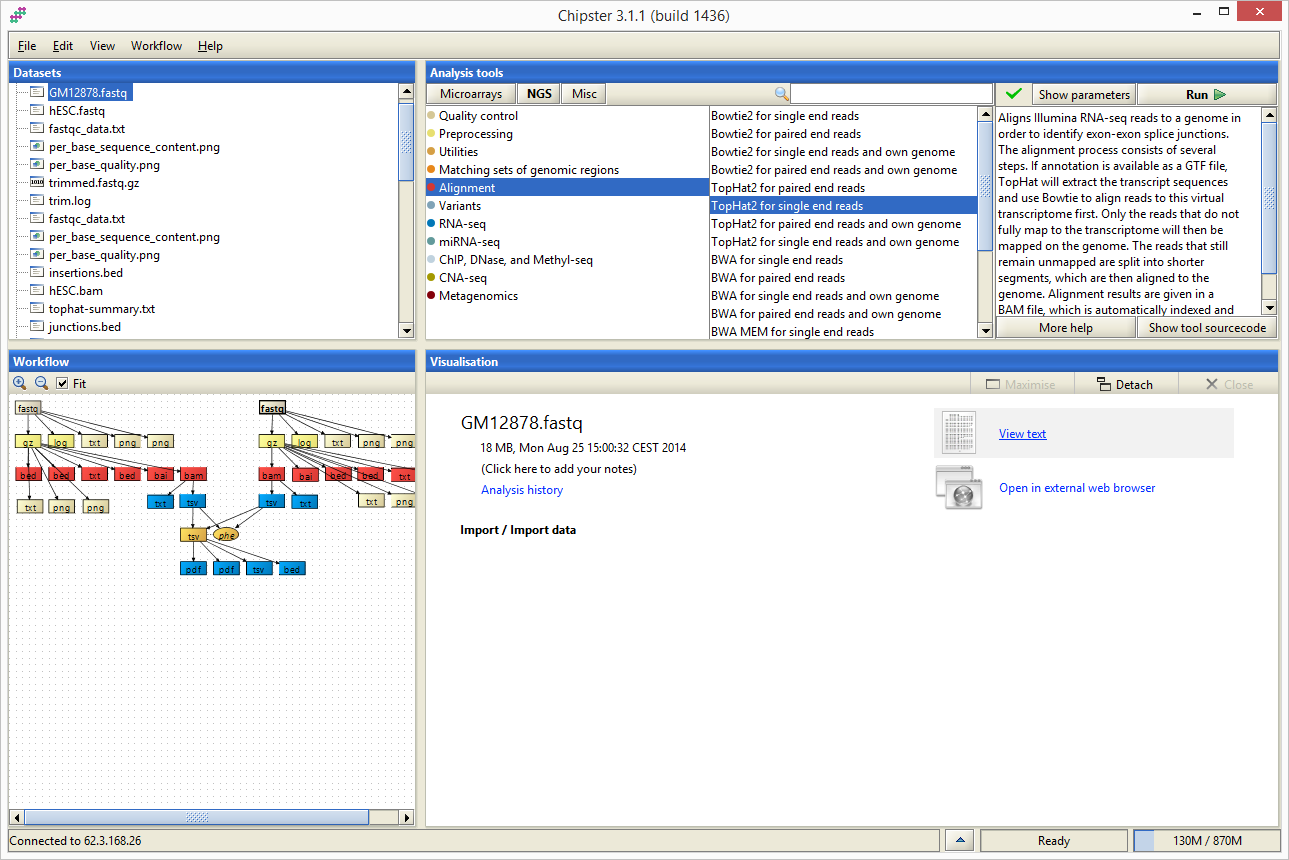
Interfejs aplikacji Chipster podzielony jest na 4 obszary: Datasets , Workflow , Analysis tools oraz Visualisation . W obszarach Datasets oraz Workflow możliwe jest śledzenie wyników analiz oraz wskazywanie plików, na których mają być uruchomione narzędzia dostępne w obszarze Analysis tools . Ostatni z obszarów, Visualisation , służy do wyświetlania informacji o wybranym pliku/wyniku oraz do wyświetlania jego wizualizacji.
Przykładowy scenariusz użycia: analiza danych RNA-seq
- Pobierz, a następnie załaduj do Chipster (używając polecenia File -> Import files ) pliki :
adrenal_1.fastq
adrenal_2.fastq
brain_1.fastq
brain_2.fastq
chr19_hg19.bed
chr19_iGenomes_GRCh37.gtf
- Mapowanie odczytów do genomu referencyjnego.Narzędzie: Alignment -> Bowtie2 for paired end reads . Przed wyborem narzędzia i parametrów zaznacz pliki adrenal_1.fastq oraz adrenal_2.fastq . Wybieramy narzędzie i parametry:
- Genome: Homo_sapiens.GRch37.75
- No 1 mate reads: plik zawierający odczyty „forward” (zazwyczaj z ‘_1’ albo ‘f’ w nazwie): adrenal_1.fastq
- No 2 mate reads: plik zawierający odczyty „reverse” (zazwyczaj z ‘_2’ albo ‘r’ w nazwie): adrenal_2.fastq
Powtórz procedurę dla plików brain_1.fastq i brain_2.fastq . Przejrzyj dostępne wizualizacje plików wynikowych. W wizualizacji genome browser należy wybrać ten sam genom, który był użyty do mapowania. Dane są dostępne dla rejonu: Chr19:3000000:3500000.
- Analiza jakości mapowania. Narzędzie: Quality Control -> RNA-seq quality metrics with RseQC . Narzędzie należy uruchomić na pliku BAM uzyskanym z mapowania w poprzednim kroku oraz załadowanym pliku chr19_hg19.bed . Przeanalizuj otrzymane wykresy oraz informacje diagnostyczne.
- Zliczanie ilości odczytów zmapowanych w obrębie genów. Narzędzie RNA-seq -> Count aligned reads per genes with HTseq-count . Uruchamiamy dla plików bam uzyskanych z mapowania paired-end z użyciem bowtie2. Parametry:
- Reference organism: Homo_sapiens.GRch37.75
- Does the BAM file contain paired-end data: yes
- Was a data produced with a strand-specific protocol: yes
- Zdefiniowanie układu eksperymentalnego. Narzędzie: Utilities -> Define NGS experiment . Uruchamiamy na plikach htseq-count.tsv z poprzedniego kroku (zaznaczyć oba przed uruchomieniem). W opcjach należy zaznaczyć kolumnę zawierającą zliczenia w obrębie plików wybranych do analizy, w naszym przypadku „count”. Po uruchomieniu powstaje tabela łącząca zliczenia z obu plików oraz plik phenodata.tsv . Plik ten należy zaznaczyć i w oknie wizualizacji wybrać Phenodata editor . Teraz należy przyporządkować próbki do grup eksperymentalnych. W naszym przypadku nie mamy replikatów, więc sample001.tsv opisujemy w kolumnie „group” jako „adrenal”, natomiast sample0002.tsv jako „brain” i naciskamy „close”.
- Testowanie statystyczne genów pod względem różnicowej ekspresji. Narzędzie: RNA-seq -> Differential expression using edgeR . Narzędzie uruchom na plikach otrzymanych w poprzednim kroku. Parametry pozostaw domyślne. Przeanalizuj wykresy diagnostyczne oraz listę genów ulegających statystycznie istotnej różnicowej ekspresji.
Gdzie szukać dalszych informacji?
Szczegółowe informacje o użytkowaniu infrastruktury PLGrid znajdują się w Podręczniku Użytkownika.
Szczegółowa dokumentacja Chipster wraz z przykładowymi sesjami znajduje się na stronie: http://chipster.csc.fi/manual/
Informacje o usługach dziedzinowych Biologia dostępne są na stronie: http://biologia.plgrid.pl/
Uzyskanie informacji/helpdesk PLGrid: dokumentacji o pomocy
Overview
Content Tools