Poniższy szablon należy odpowiednio uzupełnić.
- Układ należy zachować (z dopuszczeniem minimalnych modyfikacji).
- Opis nie powinien przekraczać 10 stron przeciętnego ekranu laptopa.
- W razie potrzeby należy założyć podstrony (na końcu z rozdziałem "Co dalej?" i odnośnikiem do kolejnego rozdziału dokumentacji).
- Język opisu - polski. W sytuacji, gdy zasadnicza dokumentacja usługi ma być po angielsku, w tym rozdziale powinny znaleźć się podstawowe informacje pozwalające zorientować się w zaletach usługi i zgrubnie w wymaganych krokach do jej uruchomienia.
- Uprawnienia do odczytu strony (Tools/Restrictions) powinny być ustawione na "Confluence-users" w trakcie pisania dokumentacji, inaczej będzie widoczna od razu dla osób niezalogowanych.
- Pytania dotyczące systemu dokumentacji: Hubert Siejkowski,
- Pytania dotyczące Podręcznika Użytkownika: Unknown User (plgfilocha).
Wstawianie odnośników do innych stron podręcznika
Przy wstawianiu linków do stron wewnętrznych Podręcznika użytkownika (np. certyfikat="Aplikowanie, rejestracja i użycie certyfikatu"' założenie konta="Zakładanie konta w portalu"; Pomoc="Gdzie szukać pomocy") należy w trybie edycji strony:
- wpisać tekst, pod który będzie podpięty link
- zaznaczyć tekst
- wstawić link (Ctrl+K lub ikona Link na pasku narzędzi)
- wybrać opcję Search z lewej strony okna Insert Link
- w pasku po prawej wpisać tytuł strony (lub zacząć wpisywać tytuł i wybrać właściwą stronę z pojawiających się podpowiedzi)
- zatwierdzić wybraną stronę opcją Insert w prawym dolnym rogu
Efekt powyższego opisu można zobaczyć klikając lewym klawiszem myszki (w trybie edycji strony) na dowolny link w tym oknie informacji. Pojawi się pole, w którym do wyboru będzie opcja Edit, którą klikamy. Pojawi się okno Edit link, z aktywnym polem Search i nazwą strony wewnętrznej podręcznika.
Przy wstawianiu linków do konkretnych sekcji (akapitów) na wybranej stronie, np. Certyfikaty Simple CA ="Aplikowanie, rejestracja i użycie certyfikatu#Certyfikaty Simple CA", należy nazwę strony wraz z tytułem sekcji, rozdzielone znakiem #, podać w opcji Advanced w okienku link. Przy wstawianiu nazwy stron istotne jest zachowanie znaków spacji pomiędzy wyrazami oraz braków spacji pomiędzy #.
LINKI ZEWNĘTRZNE
Linki zewnętrzne np. do strony PL-Grid wstawiamy w oknie Insert Link (Ctrl+K) w opcji Web Link.
Pomoc w pisaniu stron w Confluence: https://confluence.atlassian.com/display/DOC/Using+the+Editor
Krótki opis usługi
Usługa jest przeznaczona dla biologów oraz bioinformatyków.
Chipster jest implementacją popularnego środowiska zarządzania zadaniami pozwalającego na uproszczone uruchamianie analiz bioinformatycznych na zasobach obliczeniowych PLGrid. W ramach usługi szczególny nacisk położony został na udostępnienie możliwie dużej liczby narzędzi związanych z analizą danych pochodzących z eksperymentów opartych o metody wysokoprzepustowego sekwencjonowania. Wykonywanie analiz z użyciem Chipster oparte jest o wygodny interfejs dostępny w formie aplikacji Java, pozwalający na intuicyjne zarządzanie danymi, narzędziami oraz wynikami. Wbudowane moduły wizualizacji pozwalają na przejrzystą i efektywną analizę wyników.
Aktywowanie usługi
Aby skorzystać z usługi Chipster, należy mieć aktywne konto w infrastrukturze PLGrid.
Do usługi Chipster może uzyskać dostęp każdy użytkownik PLGrid, który jest użytkownikiem usługi Molecular Biology Data Analysis Toolkit. W celu uzyskania dostępu do tych usług należy wejść na stronę http://portal.plgrid.pl a następnie zalogować się podając swój identyfikator plgrid (np. plgkowalski) i hasło do portalu, po czym z górnej belki zawierającej menu wybrać opcję „Moje konto”. W prawej kolumnie ukaże się Katalog usług dostępnych dla danego użytkownika.
Z katalogu usług należy wybrać kategorię: Platforma Dziedzinowa – Biologia, a następnie „rozwiń”, ukaże się lista usług w tej kategorii, wśród nich będzie usługa o nazwie Molecular Biology Data Analysis Toolkit a tuż obok link zatytułowany „aplikuj o usługę”. Akceptacja użytkownika następuje automatycznie. Gdy rejestracja się powiedzie, usługa na liście w katalogu zyska status „aktywny”.
Pierwsze kroki
Uruchomienie usługi
Uruchomienie aplikacji Chipster możliwe jest na każdym komputerze wyposażonym w przeglądarkę internetową oraz środowisko Java Web Start. Usługa dostępna jest poprzez portal MBDAT bądź pod adresem: http://chipster.biologia.plgrid.pl. Na wskazanej stronie należy wcisnąć link launch Chipster. Alternatywnie, można również wybrać wersję aplikacji możliwą do zastosowania na komputerach o większej ilości pamięci RAM: 3 lub 6 GB. Po wciśnięciu linku pobrany zostanie plik startowy aplikacji chipster.jnlp. Pobrany plik należy uruchomić za pomocą Java Web Start. Podczas uruchamiania aplikacji nastąpi monit o zalogowanie z użyciem danych konta PLGrid.
Organizacja interfejsu
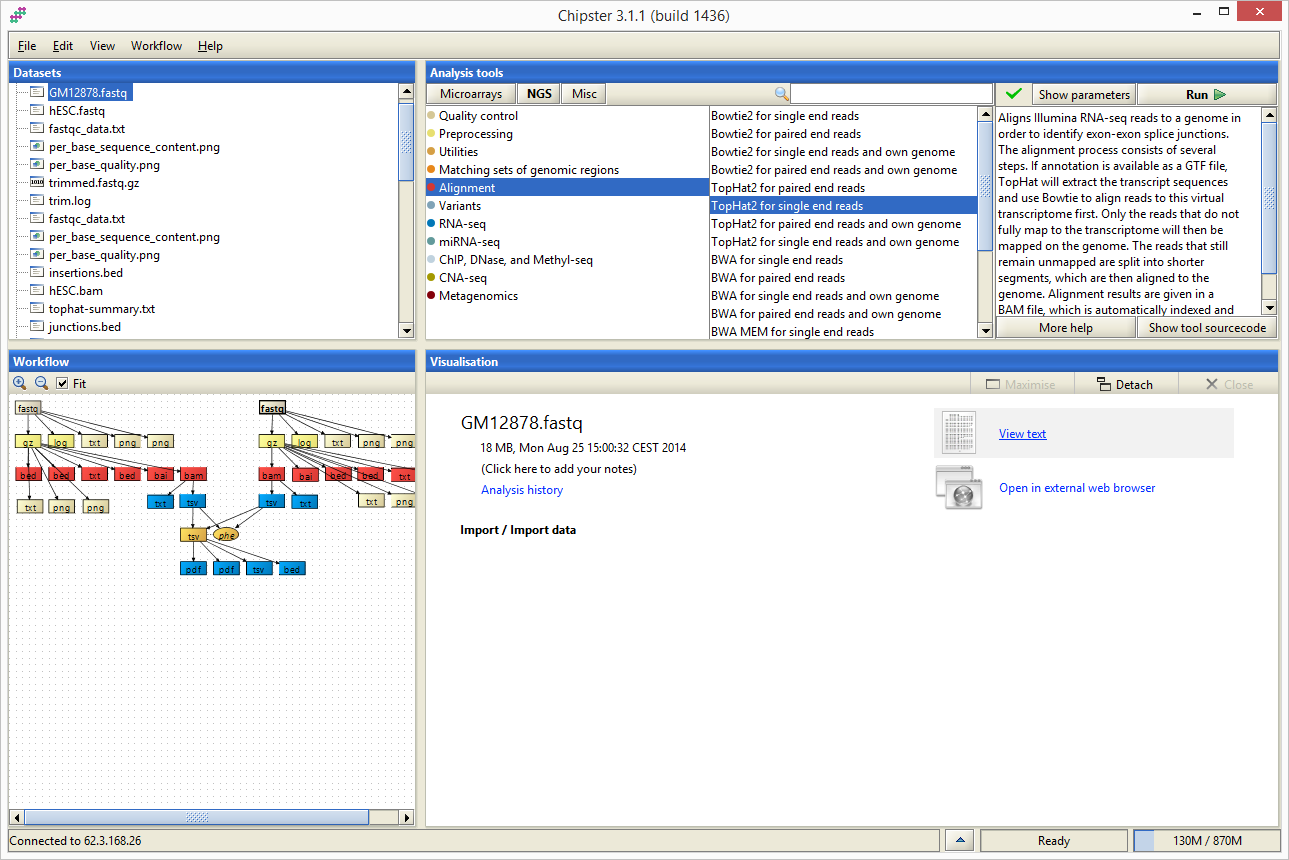
Interfejs aplikacji Chipster podzielony jest na 4 obszary: Datasets, Workflow, Analysis tools oraz Visualisation. W obszarach Datasets oraz Workflow możliwe jest śledzenie wyników analiz oraz wskazywanie plików na których mają byc uruchomione narzędzia dostepne w obszarze Analysis tools. Ostatni z obszarów, Visualisation, służy do wyświetlanai informacji o wybranym pliku/wyniku oraz do wyświetlania jego wizualizacji.
Przykładowy scenariusz użycia: analiza danych RNA-seq
- Pobierz, a następnie załaduj do Chipster (używając polecenia File -> Import files) pliki :
adrenal_1.fastq
adrenal_2.fastq
brain_1.fastq
brain_2.fastq
chr19_hg19.bed
chr19_iGenomes_GRCh37.gtf - Mapowanie odczytów do genomu referencyjnego. Narzędzie: Alignment -> Bowtie2 for paired end reads. Przed wyborem narzędzia i parametrów zaznacz pliki adrenal_1.fastq oraz adrenal_2.fastq. Wybieramy narzędzie i parametry:
- Genome: Homo_sapiens.GRch37.75
- No 1 mate reads: plik zawierający odczyty „forward” (zazwyczaj z ‘_1’ albo ‘f’ w nazwie): adrenal_1.fastq
- No 2 mate reads: plik zawierający odczyty „reverse” (zazwyczaj z ‘_2’ albo ‘r’ w nazwie): adrenal_2.fastq
Powtórz procedurę dla plików brain_1.fastq i brain_2.fastq. Przejrzyj dostępne wizualizacje plików wynikowych. W wizualizacji genome browser należy wybrać ten sam genom, który był użyty do mapowania. Dane są dostępne dla rejonu: Chr19:3000000:3500000.
- Analiza jakości mapowania. Narzędzie: Quality Control -> RNA-seq quality metrics with RseQC. Narzędzie należy uruchomić na pliku BAM uzyskanym z mapowania w poprzednim kroku oraz załadowanym pliku chr19_hg19.bed. Przeanalizuj otrzymane wykresy oraz informacje diagnostyczne.
- Zliczanie ilości odczytów zmapowanych w obrębie genów. Narzędzie RNA-seq -> Count aligned reads per genes with HTseq-count. Uruchamiamy dla plików bam uzyskanych z mapowania paired-end z użyciem bowtie2. Parametry:
- Reference organism: Homo_sapiens.GRch37.75
- Does the BAM file contain paired-end data: yes
- Was a data produced with a strand-specific protocol: yes
- Zdefiniowanie układu eksperymentalnego. Narzędzie: Utilities -> Define NGS experiment. Uruchamiamy na plikach htseq-count.tsv z poprzedniego kroku (zanzaczyć oba przed uruchomieniem). W opcjach należy zaznaczyć kolumnę zawierającą zliczenia w obrębie plików wybranych do analizy, w naszym przypadku „count”. Po uruchomieniu powstaje tabela łącząca zliczenia z obu plików oraz plik phenodata.tsv. Plik ten należy zaznaczyć i w oknie wizualizacji wybrać Phenodata editor. Teraz należy przyporządkować próbki do grup eksperymentalnych. W naszym przypadku nie mamy replikatów, więc sample001.tsv opisujemy w kolumnie „group” jako „adrenal”, natomiast sample0002.tsv jako „brain” i naciskamy „close”.
- Testowanie statytyczne genów pod względem różnicowej ekspresji. Narzędzie: RNA-seq -> Differential expression using edgeR. Narzędzie uruchom na plikach otrzymanych w poprzednim kroku. Parametry pozostaw domyślne. Przeanalizuj wykresy diagnostyczne oraz listę genów ulegających statystycznie istotnej różnicowej ekspresji.
Zaawansowane użycie
Ewentualnie jako osobny podrozdział.
Gdzie szukać dalszych informacji?
Strony zewnętrzne (jeśli są), odnośnik do helpdesku lub strony dokumentacji o pomocy.
Można też dodać sekcję "Co dalej?" ze wskazaniem (odnośnikiem) do dalszej części dokumentacji, o ile jest wymagana.
Overview
Content Tools